
































摘要
色谱系统在药物研发中具有举足轻重的作用,色谱数据完整性和合规性直接决定药品注册成败。本文是笔者基于多年药物研发经验下对色谱数据完整性理论的简浅总结,不妥之处,还望指正!
数据完整性是指数据和流程在其整个生命周期中的整体完整性、准确性和一致性。
虽然这看起来很简单,但真正以完整性和准确性生成、维护和转换数据的整个过程对于任何从事药物制造者来说都是一项具有挑战性的任务。而色谱数据完整性更是体现在色谱系统、对照品管理、样品管理、原始记录管理以及Excel验证等方面。
一、色谱系统
- 色谱工作站
色谱工作站获得的色谱数据应当可靠、安全、完整、可溯源。药物制造者应该选择、安装和使用适合其既定用途的色谱系统,并可以得到稳定的售后服务。系统应符合法规要求和GMP要求,包括但不仅限于,确保数据采集、处理和存贮符合国家法律和可追溯、清晰、同步、原始和准确的原则。鼓励采用经规范和系统验证的色谱工作站进行研究工作,但并不是说非要使用安捷伦系列、Empower、Solutions系列等软件。不过国内色谱工作站的靠谱性值得商榷。目前国内大中型药物制造从事者大部分已经采用了本地无数据储存的网络版工作站,确保了数据的安全性、可靠性和完整性。
- 验证与确认
色谱系统在进行药物制造行为前需要按照经过批准的验证方案进行验证。色谱系统验证和确认的范围和程度应根据风险管理原则确定。验证和确认应在方案中进行说明,并记录在报告中。报告中应包括文件化证据,如截屏、打印输出或其它作为验证和确认的一部分所执行检测的源数据。数据应提供证据证明系统性能一致性,以及结果可靠准确。参数(例如但不仅限于密码控制、审计追踪、访问和权限)应在验证和确认时说明并核对。如发现任何校正参数超出校准范围或不符合既定限度,则应进行根本原因分析、影响性分析和风险评估。应采取适当的CAPA。
1)新购色谱系统需要进行验证,并组织好相关文件的撰写和审评,下图是相关文件梳理。
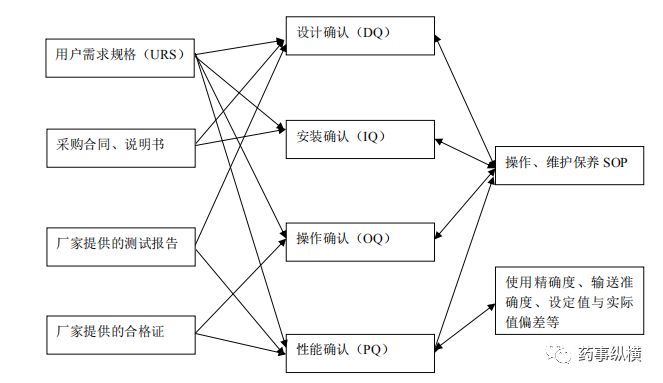
当然上述所有行为均是由总验证计划所决定的,有些色谱实验室还将操作确认和性能确认合并成OPQ或将安装确认和操作确认合并成IOQ,但一般不建议将三者合并形成IOPQ。具体详见笔者撰写的《色谱系统验证和确认理论》。
2)旧色谱系统应该约定周期性进行期间核查进行再确认,已确定系统是否满足使用。每年需经具有鉴定资格的鉴定机构对仪器进行鉴定并出具合格证。
- 访问与权限
1) Windows账户应只有一个管理员账户,管理员账户仅限于系统维护使用。对于每个实验人员应该分别有自己独立的账户和密码,每个账户仅有写入和读取权限。所有账户应该设置计算机屏幕保护,并在短时间内自动锁屏。即使管理员也不应该具备删除数据,更改系统时间的权限。
2)工作站应有标准操作程序(SOP)规定色谱系统用户组和用户的创建与删除,写明为每个用户所分配的权限。记录应保存。每个组内的用户应有适当资质可承担指定的职责和权限。用户组、用户及其权限的手工记录应与电子数据相一致。应该建立一个关于密码的SOP。工作站在无操作时应短期内锁定。即使管理员也不应该具备删除数据的权限。
- 审计追踪
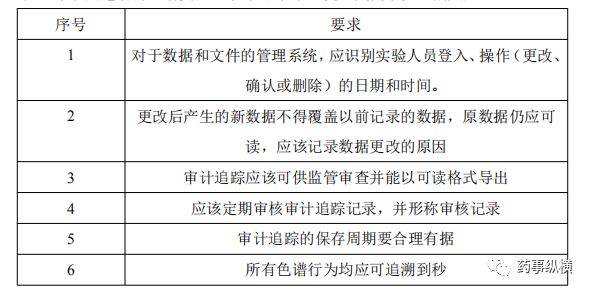
以往我们使用的ChemStation、Solution等工作站并不具备审计追踪功能,随着药物审查的日益严格,大部分色谱工作站厂家均升级了自己的工作站。目前国内各大型药企均已配备具备审计追踪功能的色谱工作站。在色谱工作安装开始即应全面激活审计追踪功能,并在色谱系统的生命周期中保持一致处于激活状态。审计追踪应根据SOP进行审核。应有证据证明对审计追踪进行了日常(每次色谱分析)和定期审核。审计追踪是元数据的一部分,应作为所有色谱数据系列一并存贮。下表是笔者对色谱实验室审计追踪的基本要求做简单总结概括:
- 签名
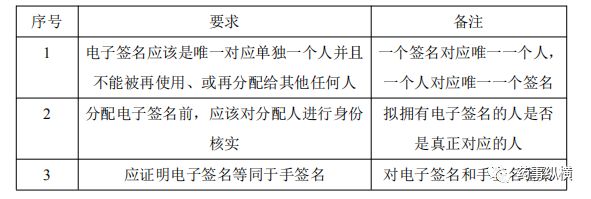
目前可以校验个人身份的方式主要有:手签名、电子签名、生物测定学以及数字签名。其中电子签名是色谱系统中常用的方式。电子签名是指一种有一个人执行、采用或批准成为与其个人的手写签名具有相同的法律效力的计算机数据的任意符号或一系列符合的编译。下表是电子签名的一般要求
- 色谱柱
色谱柱是色谱系统中的核心部分,在进行药物制造行为时应选用合适的经过批准的供应商提供的色谱柱,新收到色谱柱应该进行验收(包括柱效测试)并形成记录归档。应该建立色谱柱使用档案(因至少包括使用时间区间、用途、使用状况、最后封存液体等)。每次实验应该详细记录色谱柱信息。应该按照经批准的色谱柱再生SOP进行再生。退役色谱柱应该由相关的退役流程并贴标识统一管理。
- 溶剂及流动相
溶剂、缓冲液和流动相制备、存贮和使用应根据批准的质量标准、程序和药典。应在其经过验证的有效期内使用。详细记录配制过程并准确记录所用到化学品、试剂和其他物料的信息(至少包括厂牌、批号、级别等)。溶剂及流动相的使用应该按照SOP操作。
- 色谱方法
1)采集方法
详细记录采集方法开发过程并且对确定的采集方法进行详细的方法学验证,这些工作应由有资质有经验的人员按要求进行。经过方法学验证后的采集方法应尽可能由指定人员创建和保存在色谱系统中。采集方法不应随意进行修订,如需修订应由授权人员根据其意向目的进行批准,修改后重新进行方法学验证才可以进行产生效力的样品测定。应规定样品序列,对照液、样品溶液和空白溶液进样瓶应进行核对以确保色谱系统进样序列正确。
最好不要“试针”、“系统检查进针”或其它未规定作为样品序列一部分的进针。例外情形需要使用时,应在批准程序里清楚说明。但只有对照溶液可以用于此目的,样品是绝对不能进行“试针”检查的。最好记录进样前序列表和进样后序列表,这样可以对比进样前后的实际情况,除非色谱工作站具有此功能。每个样品序列表应单独跟踪空白对照,可以监测是否会产生干扰。应该制定系统适用性的可接受范围。
2)处理方法
所采集的结果应使用经过验证的方法进行处理。所选方法应可追溯,并反映在审计追踪里。处理方法应该符合可追溯、清晰、同步、原始和准确的原则。处理方法应该根据实际情况调整斜率灵敏度、噪声阈值、峰宽、峰面积阈值和集束因子及撇去比率的参数。一般来说色谱峰准确积分的必要条件是所有峰能完全分离。如果必须从未分开的峰中获得定量数据,则实验室应制订明确的方针说明如何对这些峰进行积分。其中应包括阐明何时可使用不同函数对未分离的峰进行积分。
如撇去设置、通过设置峰谷比和峰高比来调整等。一般来说是不建议手动积分的,非要进行手动积分应该按照建立的手动积分SOP进行并保存好记录。自动积分和手动积分的谱图必须同时保存。手动积分应该按最差情况积分。积分过程中的平滑处理是要经过论证处理的。色谱系统应该独立保存每次数据处理后的结果并且保证每次结果可读。同一样品序列应该使用同一个处理方法。
- 数据管理
所有数据在灾难(如仪器故障、病毒、硬件或软件故障和断电)发生时可恢复。所获取的色谱数据应可追溯、清晰、原始和准确。数据应根据程序备份,记录应保存作为证据。应特别注意确保单机系统数据备份要频繁,以防止数据遗失。数据应安全存贮,包括对数据访问进行控制。备份数据应按指定时间间隔随机抽取进行恢复和验证。完整数据应保存适当时长,期间可进行数据核查、注册申报或其它。
二、对照品管理
对接收的对照品需进行验收(对照品名称、数量、有效期、供应商、外观状态、纯度复核及对照品证书),合格后进行登记入账,并按说明书或指定的储存条件严格储存。一般按标准品的规定贮存期限执行,没有期限的原则上化学提纯物标准品为3年(生物试剂和不稳定的以6~12个月为宜)。对照品纯度标定在在验收时一并进行。
实验人员根据项目,领用对照品,对照品管理员根据标准品存放位置,寻找标准品交于实验人员。实验人员初步验收对照品后,填写领用记录,记录应至少包括(对照品名称、纯度、厂家、规格、批号、领用时间、领用人、用途、领用前质量和领用后质量等信息)。对照品的第一个开封者,应将对照品的开瓶日期、纯度、有效期标注于标准品瓶上,并签名。色谱实验室用到的所有对照品都应该有合理的有效期,对照品的有效期应基于厂家提供、历史数据的总结或稳定性研究的结果。对照品过期或出现异常无法使用时,应提出销毁申请,不能私自处理。
由对照品管理员记录。记录内容至少包括品名、规格、数量、销毁原因、购入单位、购入日期、申请人、批准人。对照品溶液的配制过程应该详细记录并标注配置信息、储存条件及有效期。所用官方、第二或工作对照品应可通过保存的记录追溯其采购、配制、存贮和使用情况。
三、样品管理
样品进入色谱实验室应该填写样品送检单(送检单应至少记录送检样品名称、送检数量、批号、送检人、结构式、储存条件、外观及简单物理性质等)。应录入适当记录,确保样品详细信息和分析可追溯。样品也应简单记录使用情况,例如使用前质量、使用后质量、使用日期、用途使用人等。
四、原始记录管理
原始记录管理见笔者撰写的《依6M分析法形成的药物科研实验室中的原始记录》及《药物分析测定中修约规则理论》。在此不做赘述。
五、Excel验证
色谱实验中最常用的计算辅助工具就是Excel,Excel也是色谱数据完整性必不可少的条件,因此保证Excel数据的可追溯、清晰、原始和准确是十分必要的。使用Excel计算一些关键数据(产品放行、关键控制指标)前,需要对Excel进行验证。对于色谱实验室来说只需要验证用Excel软件创建的电子表格中的内容。主要包括计算公式、数据引用、数据检查、数据格式、数据精度等。具体验证理论见笔者撰写的《色谱实验室中Excel数据可靠性验证理论》。
结语
以上是笔者总结个人经验并结合国外理论对色谱实验室数据完整性相关要求的漫谈。保证色谱实验室的数据可追溯、清晰、原始和准确是一项及其复杂繁重的工作。任何理论性指导原则仅是我们从事色谱行为的基本,只有在实验中不断积累经验,养成保证色谱数据完整性的习惯,才能真正做出可追溯、清晰、原始和准确的色谱数据。
PS:由于笔者经验尚浅,欢迎广大专家批评指正,不喜勿喷!
参考文献
【1】 Data Integrity and Compliance With CGMP Guidance for Industry
【2】 Validation of computerised System-Core document PA/PH/OMCL
【3】 GOOD CHROMOTOGRAPHY PRACTICES
【4】 21 CFR part 11 complete guide to international computer validation compliance for the pharmaceutical industry
相关阅读:
<END>



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论