



























2022年已经过去,据药融云统计,在这一年中美国FDA一共批准了37款新药上市,其中包含22个新分子实体和15个新生物制品申请。这也是2017年以来,美国FDA批准新药上市数量最少的一年。在所有批准的药物中,抗癌药依旧占主体,多个重磅抗癌疗法陆续上市,给患者带来了更多选择。因篇幅有限,将分为两篇来发布。此前已经发布了前18款(《2022年美国FDA批准新药上市的37款药物盘点(一)》),此篇为后19款。
2022年美国FDA批准新药上市的37款药物
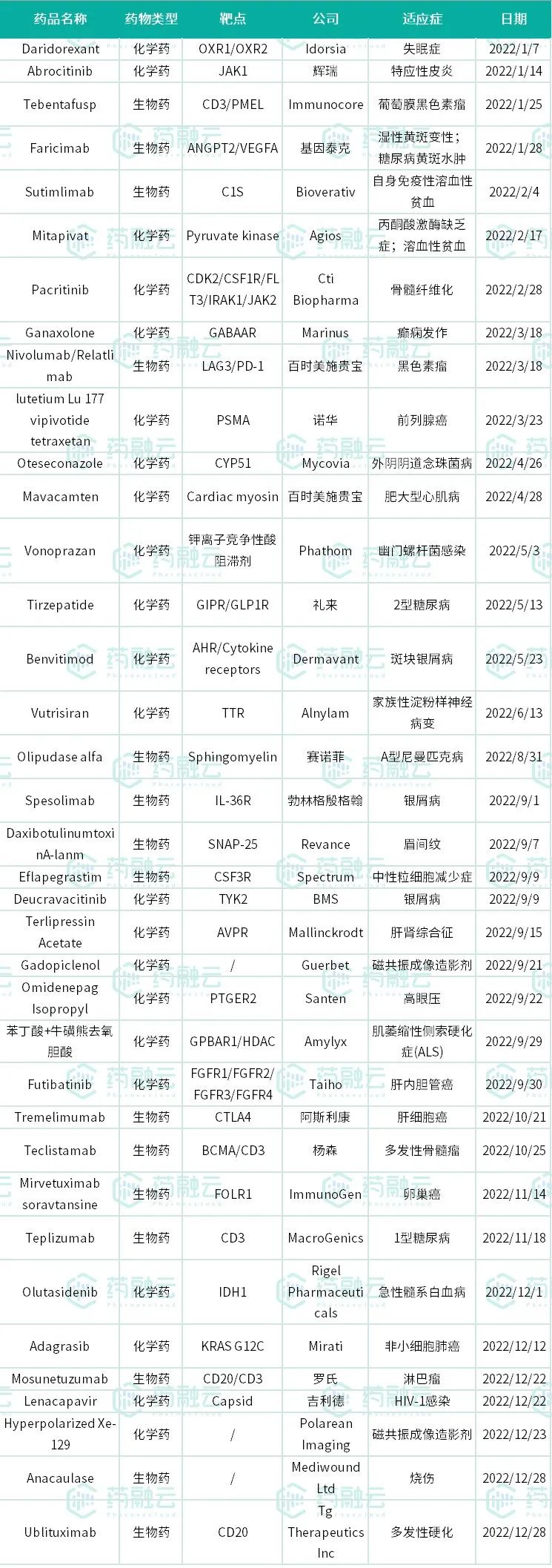
数据来源:药融云企业版数据库综合查询
资源下载
“药融云公众号(yrydata)”后台回复关键词“FDA2022”,即可获取《2022年FDA批准的新药》。
- 19.Daxxify(daxibotulinumtoxinA)
2022年09月08日(田纳西州纳什维尔)Revance Therapeutics宣布,美国FDA已批准Daxxify(DaxibotulinumtoxinA),用于暂时改善成人的中度至重度眉间纹。DAXXIFY™是第1个也是唯一1个通过肽交换技术™(PXT)稳定的神经调节剂,不含人血清白蛋白和动物成分。FDA此次批准新药上市是基于该药包含超过2700名参与者,接近4200次治疗的3期临床试验项目的结果。在关键性临床试验中,74%的受试者在接受治疗第4周,依据研究者和患者的评估,达到眉间纹改善两级以上的水平。中位疗效持续时间为6个月。最早疗效可在接受治疗1天后出现。Daxxify表现出良好的安全性和耐受性,在临床试验中未报告严重治疗相关不良事件。
- 20.ROLVEDON(eflapegrastim-xnst)
2022年09月09日,Spectrum Pharmaceuticals,Inc.宣布,美国FDA批准ROLVEDON™(eflapegrastim-xnst)注射以减少感染的发生率(治疗粒缺,同时作为粒缺伴发热的支持药物或者感染预防药物)。ROLVEDON™(eflapegrastim-xnst)注射液是20多年来首款获FDA批准新药上市的新型长效粒细胞集落刺激因子(G-CSF),通过结合在粒细胞祖细胞上表达的G-CSF受体来刺激其增殖过程,使其最终在骨髓中产生功能性活化的嗜中性粒细胞,用于治疗化疗引起的中性粒细胞减少症。
ROLVEDON的BLA得到了来自两项相同设计的3期、随机、开放标签、非劣效性临床试验ADVANCE和RECOVER的数据的支持,该试验评估了ROLVEDON在643名早期乳腺癌患者中用于治疗中性粒细胞减少症的安全性和有效性由于骨髓抑制化疗。在这两项研究中,ROLVEDON在严重中性粒细胞减少症(DSN)的平均持续时间中证明了预先指定的非劣效性(NI)假设以及与pegfilgrastim相似的安全性。ROLVEDON在两个试验中的所有四个周期(所有NI p<0.0001)的平均DSN也证明了非劣于pegfilgrastim。
- 21.Sotyktu(Deucravacitinib,氘可来昔替尼)
2022年09月09日,百时美施贵宝(BMS)宣布,Sotyktu™(Deucravacitinib,氘可来昔替尼)治疗斑块状银屑病的新药上市申请已获FDA批准。Sotyktu是第一款获FDA批准的TYK2抑制剂,FDA本次批准是基于2项关键III期临床试验(POETYK PSO-1和POETYK PSO-2)的积极结果。两项试验均为全球性、多中心、双盲、随机、安慰剂和阳性药物对照研究,分别纳入了666例患者和1020例患者,旨在评估氘可来昔替尼对比阿普米司特(apremilast)和安慰剂治疗中重度斑块状银屑病成人患者的疗效和安全性。共同的主要终点为第16周时银屑病面积与严重性指数(PASI)评分改善75%以上和sPGA 0/1(静态医生总体评估皮肤症状完全清除/几乎完全清除)的患者比例。
数据显示,第16周时,与安慰剂组和阿普米司特组相比,氘可来昔替尼组PASI评分改善75%以上和sPGA 0/1的患者比例显著增加。试验还达到了所有次要终点,氘可来昔替尼在症状负担和生活质量测量值上表现出显著且具有临床意义的改善。此外,氘可来昔替尼耐受性良好,因不良事件导致的停药率低。
- 22.Terlivaz(Terlipressin)
2022年09月14日,Mallinckrodt plc宣布美国食品药品监督管理局(FDA)批准Terlivaz®(Terlipressin)特利加压素注射剂,改善肝肾综合征(HRS)成人的肾功能迅速下降。Terlivaz是第1个也是唯一1个获得FDA批准新药上市的产品,用于改善肝肾综合征(Hepatorenal Syndrome,HRS)成人的肾功能,肾功能迅速下降,HRS是一种需要住院治疗的急性和危及生命的疾病。FDA的批准部分基于3期CONFIRM试验的结果,该试验是有史以来规模最大的前瞻性研究(n=300),旨在评估特利加压素在HRS 1型(HRS-1)患者中的安全性和有效性。美国和加拿大。
CONFIRM试验达到了验证HRS逆转的主要终点,定义为肾功能改善、避免透析和短期存活(p=0.012)。1为实现经验证的HRS逆转,患者必须在第14天或出院前连续两次血清肌酐(SCr)值≤1.5 mg/dL,至少相隔两个小时。为纳入主要疗效终点分析,患者必须在达到经验证的HRS逆转后至少10天存活且未进行肾脏替代治疗(例如透析)。初步结果在AASLD年会The Liver Meeting®2019的最新会议上公布。结果还于2021年3月发表在《新英格兰医学杂志》上。CONFIRM试验是在2021年AASLD肝肾综合征指南中发布的更新诊断标准和术语之前完成的。
与安慰剂相比,在至少4%接受Terlivaz治疗的患者中,最常见的不良反应是19.5%(n=39)的患者报告的腹痛(vs.6.1%;n=6),报告的恶心率为16%(n=32)患者(vs.10.1%;n=10),15.5%(n=31)患者报告呼吸衰竭(vs.7.1%;n=7)13%(n=26)报告腹泻的患者(与7.1%;n=7)和12.5%(n=25)的患者(与5.1%;n=5)报告了呼吸困难。
- 23.Elucirem(Gadopiclenol)
2022年9月21日,FDA经过优先审查批准了Guerbet公司的Elucireem™新药上市,这是一种用于对比增强磁共振成像(MRI)的新型大环钆造影剂。该产品用于检测和可视化中枢神经系统(大脑、脊柱和相关组织)和身体(头颈部、胸部、腹部、骨盆和肌肉骨骼系统)中的血管异常病变。Gadopiclenol是Eucirem™的活性物质,有两个水分子交换位点,用以增加弛缓性和对比度,与其他非特异性GBCA(大环钆基对比剂)相比,使用Gadopiclenol的剂量只有常规钆剂量的一半,缓解了从业者对放射性暴露的担忧。
- 24.Omlonti(Omidenepag Isopropyl)
2022年9月22日,参天公司(Santen Inc.)和UBE Corporation(UBE)宣布,美国FDA批准OMLONTI®(omidenepag isopropyl ophthalmic solution)0.002%奥米替尼异丙基滴眼液用于降低原发性开角型青光眼或高眼压患者的眼压升高(IOP)的新药上市申请。OMLONTI®在三项随机对照临床试验中对开角型青光眼或高眼压患者进行了评估,平均基线IOP为24-26 mm Hg。
在所有三项研究中,双盲治疗持续时间均为三个月。第三项研究包括在3个月的双盲治疗期之后的9个月的开放标签治疗期。在三项研究中,观察到所有治疗组的眼压降低。在OMLOTI®臂中,所有三项研究的眼压降低范围为5-7 mm Hg。噻吗洛尔和拉坦前列素组的相应降低分别为5-7 mm Hg和6-8 mm Hg。
- 25.Relyvrio(sodium phenylbutyrate+taurursodiol,苯丁酸+牛磺熊去氧胆酸)
2022年09月29日,美国FDA宣布,批准了Amylyx制药公司开发的Relyvrio(sodium phenylbutyrate/taurursodiol)苯丁酸钠和牛磺酸二醇口服固定剂量配方,用于治疗肌萎缩侧索硬化(ALS)。此次获批新药上市是基于一项入组了137例ALS患者的2期临床试验获得的积极数据。试验达到其主要疗效终点,即根据修订的ALS功能评定量表测量,在6个月随机化阶段结束时,接受Relyvrio治疗的ALS患者运动功能下降显著减缓。
- 26.Lytgobi(Futibatinib)
2022年10月30日,日本大鹏药品(Taiho Pharmaceutical)和子公司Taiho Oncology宣布,美国FDA已加速批准Lytgobi(Futibatinib)片剂新药上市,用于治疗携带FGFR2基因融合或其它重排的不可切除、局部晚期或转移性肝内胆管癌经治成人患者。LYTGOBI的批准是基于FOENIX*-CCA2试验的初步分析结果,这是一项全球2期开放标签试验,评估了103名具有FGFR2基因重排(包括融合)的不可切除、局部晚期或转移性iCCA患者。在这项试验中,患者每天口服一次LYTGOBI,剂量为20mg,直至疾病进展或出现不可接受的毒性。试验结果显示,futibatinib达到42%的客观缓解率,中位缓解持续时间为9.7个月,72%的患者缓解持续时间超过6个月。
- 27.Imjudo(Tremelimumab)
2022年10月21日,美国FDA官网显示,阿斯利康(AstraZeneca)公司开发的抗CTLA-4抗体Imjudo(tremelimumab)新药上市申请已经获得批准,与抗PD-L1抗体Imfinzi(durvalumab)联用,治疗不可切除的肝细胞癌患者,为肝癌患者提供了一种由双重免疫检查点抑制剂构成的全新免疫组合疗法。
Tremelimumab和Imfinzi组成的联合用药方案(阿斯利康将其称为STRIDE方案)对于肝细胞癌的疗效和安全性得到了临床研究积极结果的支持,在一项针对不可切除的肝细胞癌患者的3期临床试验中,研究者首先对患者进行一次tremelimumab和Imfinzi的联合用药,然后每隔4周进行Imfinzi的单药治疗,这一给药方案旨在刺激T细胞激活的同时,减少CTLA-4抗体的毒副作用。该试验结果显示,接受这一联合用药方案治疗的患者,与活性对照组相比死亡风险降低了22%(HR=0.78,96.02%CI:0.65-0.93,p=0.0035),并且总生存期、客观缓解率、3年后患者的存活率等多项指标均显著优于对照组患者。
- 28.Tecvayli(Teclistamab)
2022年10月25日,强生(Johnson&Johnson)集团旗下杨森(Janssen)公司宣布,美国FDA已经加速批准同时靶向B细胞成熟抗原(BCMA)和CD3受体的双特异性抗体Tecvayli(Teclistamab)新药上市,用于治疗复发/难治性多发性骨髓瘤(RRMM)成人患者。Teclistamab同时靶向BCMA与T细胞表面CD3受体的双特异性抗体,作为皮下治疗给药。这种现成(或即用)疗法使用创新科学,通过将CD3阳性T细胞募集到表达BCMA的骨髓瘤细胞附近,激发T细胞杀伤肿瘤细胞。它为患者提供了一款现货型、皮下注射的治疗选择。
- 29.ELAHERE(Mirvetuximab soravtansine)
2022年11月14日,ImmunoGen Inc.宣布美国FDA加速批准其抗体偶联药物(ADC)ELAHERE™(mirvetuximab soravtansine-gynx)的新药上市申请,用于治疗叶酸受体α(FRα)阳性、铂耐药上皮性卵巢癌、输卵管癌或原发性腹膜癌的成年患者,这些患者之前接受过1到3种全身治疗方案。
ELAHERE是一款的抗体偶联药物(ADC),包含叶酸受体α结合抗体、可裂解接头和美登木素生物碱有效载荷DM4(the maytansinoid payload DM4),DM4是一种有效的微管蛋白抑制剂,旨在杀死靶向癌细胞。卵巢癌患者对含铂疗法产生耐药性是成功控制疾病的一个重大挑战。
FRα是叶酸受体家族的一员,它以高亲和力与叶酸结合,导致它们被内吞摄入细胞内。之前的研究表明,FRα在76-89%的上皮卵巢癌和35-68%的三阴性乳腺癌中高度表达。使FRα成为引人关注的药物靶点。而且,FRα介导的信号通路能够影响肿瘤细胞的分裂和迁移,因此抑制FRα还可能产生一定程度的直接抗癌活性。
- 30.TZIELD(teplizumab)
2022年11月18日,Provention Bio,Inc.生物制药公司宣布美国食品药品监督管理局(FDA)批准了TZIELD(teplizumab-mzwv),用于延缓8岁及以上患有2期1型糖尿病(T1D)的成人和3期1型糖尿病(T1D)儿童患者的发作。该疗法的新药上市获批是基于一项随机、双盲、设置对照的临床试验。有76名1型糖尿病患者参与了该试验,并被随机分为两组,一组接受teplizumab治疗,另一组则接受安慰剂。治疗一共维持了14天,而在随后的随访中,治疗组的44名患者里,有45%最后出现1型糖尿病的进展,而对照组的数字则高达72%。此外,治疗组患者出现疾病进展的时间约为50个月,而对照组的数字仅为25个月。这些数据表明teplizumab的治疗可以显著延缓1型糖尿病的病情进展。
- 31.Rezlidhia(Olutasidenib)
2022年12月01日,Rigel Pharmaceuticals公司宣布,美国FDA已经批准异柠檬酸脱氢酶1(IDH1)选择性抑制剂Rezlidhia(olutasidenib)的新药上市申请,用于治疗复发或难治性(R/R)急性髓系白血病(AML)患者,这些患者具有易感异柠檬酸脱氢酶-1(IDH1)突变。
Rezlidhia的的批准基于针对147名患有复发性或难治性AML患者(都有IDH1突变)的临床研究。疗效的评估基于完全缓解率(CR)加上完全缓解伴部分血液学恢复(CRh)的比率、CR+CRh的持续时间。CR+CRh率为35%,CR+CRh的中位时间为1.9个月,CR+CRh的中位持续时间为25.9个月。
- 32.Krazati(adagrasib)
2022年12月12日,美国FDA宣布加速批准Mirati公司KRAS G12C抑制剂Krazati(adagrasib),用于治疗携带KRAS G12C突变的局部晚期或转移性非小细胞肺癌(NSCLC)患者,这些患者先前至少接受过一种全身性疗法。这是FDA批准的第二款直接抑制KRAS突变体活性的靶向疗法。
FDA此次的批准新药上市是基于adagrasib在支持注册的2期临床试验KRYSTAL-1中的队列结果。在携带KRAS G12C突变的晚期NSCLC患者中,adagrasib达到43%的客观缓解率(ORR)和80%的疾病控制率(DCR)。
- 33.Lunsumio(Mosunetuzumab)
2022年12月22日,罗氏(Roche)旗下基因泰克(Genentech)宣布,美国FDA已批准Lunsumio(Mosunetuzumab-axgb)新药上市,用于治疗经过两种或多种前期系统治疗后复发或难治的滤泡性淋巴瘤(FL)成年患者。此次FDA的批准是基于2期GO29781研究的积极结果,该试验结果显示出Lunsumio对于复发或难治滤泡性淋巴瘤患者具有较高且持久的应答率。在接受Lunsumio治疗的患者中,80%的患者出现了客观反应(包括完全反应和部分反应),大多数患者至少18个月内保持了反应(57%)。应答者的中位应答持续时间接近2年。60%的患者获得完全反应(CR)。
- 34.Sunlenca(Lenacapavir)
2022年12月22日,吉利德科学(Gilead Sciences)宣布FDA批准其药品Sunlenca(Lenacapavir)注射液和片剂,用于联合其他抗逆转录病毒药物治疗多重耐药人类免疫缺陷病毒(HIV)感染的成人患者。Sunlenca是首个基于衣壳抑制剂的HIV治疗选项,该药的新药上市批准主要基于2/3期临床试验CAPELLA数据的支持,该试验评估了Sunlenca联合优化背景抗病毒疗法治疗方案在接受过大量治疗的多重耐药HIV患者中的应用。
在显著未满足医疗需求的患者人群中,83%(n=30/36)接受优化背景方案的基础上加用Sunlenca的受试者在第52周时达到病毒载量检测不到的标准(<50拷贝/毫升)。此外,CAPELLA受试者的CD4阳性细胞计数平均增加82个细胞/微升。《新英格兰医学杂志》于今年5月发表了CAPELLA研究的主要结果。
- 35.Xenoview(Hyperpolarized Xe-129)
2022年12月28日,FDA批准Polarean Imaging开发的Xenoview(Hyperpolarized Xe-129)新药上市,该药是一种用于磁共振成像(MRI)的超极化造影剂,用于评估成人和12岁及以上儿童患者的肺通气量。当吸入超极化氙Xe 129气体时,可以使用具有多核能力的MRI扫描仪对整个通气肺中的超极化氙Xe 129分布进行成像。
- 36.NexoBrid(Anacaulase)
2022年12月28日,MediWound公司宣布美国FDA已批准其新药NexoBrid(anacaulase-bcdb),用于移除有深度部分皮层烧伤和/或全皮层烧伤的成人患者身上的焦痂。该药物的新药上市申请基于一系列临床前研究,以及8项临床试验的数据,其中包括一项关键的美国3期临床试验。研究表明与对照相比,该药物可以达到95%及以上的焦痂移除,达到主要临床终点。
此外,该疗法也达到了一系列次要临床终点,包括焦痂移除时间更短、需要手术移除焦痂的比例更低,以及与手术和非手术的标准护理方式相比,失血更少等。在安全性上,与标准护理方式相比,该疗法也达到了非劣效性的临床终点。总体上看,该疗法安全且耐受良好。
- 37.BRIUMVI(Ublituximab)
2022年12月28日,TG Therapeutics,Inc.宣布,美国FDA已批准BRIUMVI™(ublituximab-xiiy)用于治疗复发性多发性硬化症(RMS)的各种形式新药上市,包括成人的临床孤立综合征、复发缓解型疾病和活动性继发性进展性疾病。Briumvi的获批基于ULTIMATE I和ULTIMATE II两项为期96周的3期试验研究,试验在1,094名复发型MS患者中进行。接受Briumvi治疗的患者的年化复发率(主要终点)分别为0.076和0.091,而特立氟胺组分别为0.188和0.178。
想要解锁更多药物研发信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、申报审批情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!

—END—









 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论