































在“新药早期开发中关键理化性质考量-溶解度”文中,简单探讨了作者对于平衡溶解度中“平衡”两字的理解,接着从溶解度计算公式以及晶体药物溶解的简单模式图出发,解释了溶解的过程以及影响化合物溶解的关键控制指标:化合物熔点及LogP。上文一经发出,感觉要说的话还没有说完,有种意犹未尽之感,于是乎“新药早期开发中关键理化性质考量-溶解度、溶出与体内吸收”发车。
溶解度、溶出与吸收:
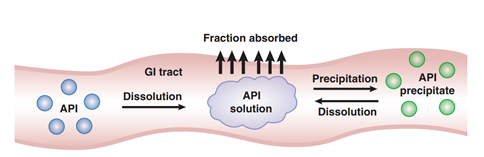
图1 药物体内过程图(来源于文献1)
图1描述了药物制剂在人体胃肠道中吸收的过程,揭示了溶解度和溶出度如何去影响口服固体制剂的体内吸收。药物必须在溶液中才能被吸收,吸收以后才有可能发挥作用。药物制剂溶出将推动API的初始溶解,从图2可以看到即使对于一个普通的口服固体制剂其溶出亦相当的复杂,药物制剂的崩解成初级粒子(K2),初级粒子的解聚(K3),解聚API粒子的溶出(K5),这些步骤都可以限制药物的溶出。
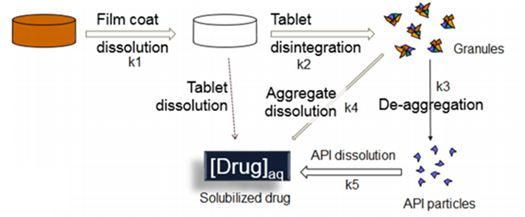
图2 普通口服固体制剂溶出图(来源于文献2)
再回到图1,在胃肠道介质中的药物溶解度将推动潜在的沉淀发生,比如弱碱性化合物成盐后,溶出速率的提高增加的溶解度,即在胃肠道形成过饱和溶液,可是药物溶液转运到碱性的小肠,极有发生沉淀可能,进而影响药物的吸收,如图3所示。
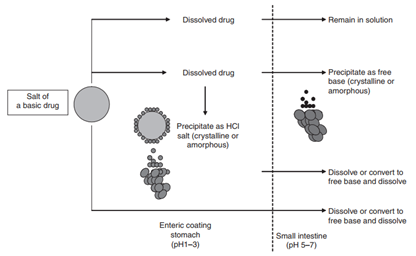
图3 碱性药物成盐后在胃肠道的溶出过程(来源于文献3)
其实,我们抛开溶解度和溶出度这些概念,聚焦在物吸收的那个狭小的部位,药物吸收不过是一个很简单的跨越细胞的过程,而这个过程与此时细胞两侧的药物浓度有关(因为大部分化合物以被动转运为主)。
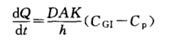
被动转运是药物自发的由高浓度一侧向低浓度一侧扩散,无需消耗能量,可以用Fick’s方程来描述。Fick’s方程中是扩散速度,D是扩散系数,A膜表面积,K是药物的分配系数,h是膜厚度,CGI为胃肠道药物浓度,Cp为血浆中药物浓度。
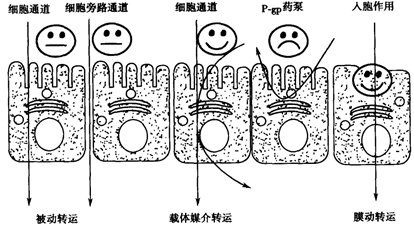
图4 药物跨膜转运机制示意图(来源于文献4)
从Fick’s方程可知,对于一个药物分子来说,化合物的扩散系数D,化合物的分配系数K以及吸收部位的膜表面积A,膜厚度h是一致的,而血浆中药物浓度Cp,可以随着药物进入血液循环进入全身各处,故血浆中药物浓度Cp可以忽略不计。仅剩余一个参数就是胃肠道药物浓度CGI。当然CGI的高低离不开化合物的基本理化性质-溶解度和溶出对于其的影响。
最后再次回到图1,溶解的药物在胃肠道部位,形成一定浓度的药物溶液,化合物将跨膜入血,即药物吸收。
以上描述了一个药物制剂如何溶出,如何形成胃肠道溶液,如何进入血液循环的过程,同时也看到了药物溶解度及溶出在药物吸收中所发挥的巨大作用,不知道大家会不会有这样的疑问:
平时我们一般测定化合物的平衡溶解度,少则24h,多则48h、72h,而药物在胃肠道(主要指代小肠药物主要吸收部位)不过区区几个小时,这个时间的差距有点过大,24h的平衡溶解度的数值如何指导胃肠道几小时的药物吸收?
当然这是我的疑问,一直还未有清晰的答案。
平衡溶解度更像是一个晶体药物溶解的程度,达到稳态的一个数值,可能不同试验来测定,略有不同,但是不会有质的差距,易于进行表征。诚然,溶解的程度之深,不代表溶解速率之快,溶出似乎可以用来表明这个药物制剂在体内溶出快慢的情况,更加有意义去与体内药物的吸收去建立联系。比如,药物可以在体外漏槽条件下,15min溶出85%以上,这个药物制剂在体内似乎还未经历胃排空,已经基本上变成了溶液制剂。当然,抛开化合物的溶解性来谈溶出也是不对。
口服固体药物制剂后,药物在胃肠道内经历崩解、分散、溶出过程才可通过上皮细胞膜吸收,如果药物为水溶性,其崩解后可立即进入分散、溶出过程,能够迅速被吸收,则崩解是水溶性药物吸收的限速过程。对难溶性药物而言,药物从固体制剂中溶出的速度很慢,尽管崩解分散过程很快,其吸收过程往往受到药物溶出速度的限制,溶出是难溶性药物吸收的限速过程。在这种情况下,药物在胃肠道内的溶出速度直接影响药物的起效时间、药效强度和作用持续时间。可见,药物溶解性之于溶出的重要性。
图5描述药物从固体药物颗粒中溶出过程。在溶解固体的表面上存在一个宽度为(h)的未搅拌水层。在未搅拌的水层两侧形成了浓度梯度,驱动溶出。Noyes Whitney方程很好地用参数把药物溶出与溶解建立了联系。
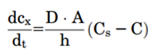
药物制剂在溶出介质的扩散速率是未搅拌层的浓度梯度、扩散层的厚度(h)、固体与溶出介质的接触表面积(A)和药物水中扩散速率(D)所决定。而未搅拌层的浓度梯度由溶解固体表面上的药物浓度(通常假定为药物的平衡溶解度,Cs)与溶出介质中药物浓度(Cp)之间的差值构成。一般溶出介质的体积要符合药物完全溶解所需介质体积的3倍以上,此为漏槽条件,这样是为了模拟药物在胃肠道吸收以后,迅速随着血入全身,Cp被稀释,可以忽略不计。Noyes Whitney方程可以简化成下式,这样就形成溶解度-溶出的关系。
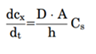
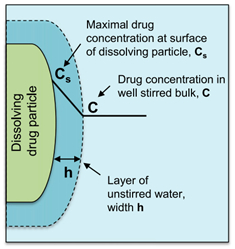
图5 描述药物从固体药物颗粒中溶出过程(来源于文献5)
溶解度限制药物体内吸收:
药物在缓冲溶液的溶解度是一项非常重要的性质,它不仅影响口服给药后药物吸收的可能性,而且还影响在实验室中测试的容易程度。对于上面的描述进行总结,药物溶解度是一种平衡量度,溶出则是速率的度量,肠道转运时间是有限的,药物在胃肠道吸收部位形成一定浓度的药物溶液且停留一定的时间才能使吸收最大化。
药物的溶出速率在口服给药后尤其重要,如果溶出率低,即使药物溶解度极佳,药物仍旧可能存在吸收问题,因为溶出的时间延长可能完美错过了吸收部位而造成溶出无效,这样就造成了溶出限制药物吸收。类似地,即使溶出速度相对较快,如果平衡溶解度低,溶液中可用于吸收的药物量也不可能支持足以吸收整个药物剂量,这样就造成了溶解度限制药物吸收。
口服药物分类系统(DCS)在生物药剂学分类系统(BCS)的基础,如图6,所示,创造性的引入以下概念:(1)人类空腹肠道溶解度的估计值(例如,通过使用FaSSIF)作为体内溶解度的主要衡量标准,可用于预测范围人体吸收。小肠作为人体胃肠道吸收的主要部位,小肠部位的环境对于药物的溶解似乎更加重要,模拟肠液很好的模拟了小肠中的胃肠道环境,包括胃肠道酸碱性环境以及胆汁等可能存在增溶的物质;(2)溶解度限制吸收剂量(SLAD)概念,创造性的划分了溶解度及溶出限制药物吸收的分界线。在制剂早期开发策略上,提供了指引,针对药物的性质,可以制定化合物不同的固态开发策略。当然,DCS仅仅是一预测性的工具,随着对于其认识的加深,究竟能起到多大的作用,需要在更多的项目中进行应用与验证,与最终的体内情况进行比对,更进一步的指导其他项目开发。从实践中来,到实践中去。
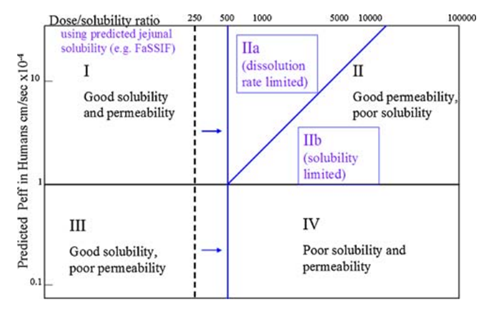
图6 BCS与DCS(来源于文献6)
小结:在新药开发中,既要看到溶解度这一药物理化性质的重要作用,更要看到其在药物体内吸收中真正发挥到的巨大影响。知其然,知其所以然。药物的平衡溶解度主要是溶解度的程度,在药物的体内吸收中,主要还是要关注药物制剂溶出的速度。当然,溶解的程度与溶出的速度都能限制药物口服吸收。孔子说,因材施教。我们针对药物吸收情况,也可以“因限施制”。
一直在路上,还不能停歇!
以上拙笔,望不惹得大家们哄堂大笑,如有不妥之处,还请留言;如有大为收获之处,还请点赞,转发!!!
推荐阅读:
《新药开发之多晶型理化性质底层逻辑》
参考文献:
1.Pharmaceutical Amorphous Solid Dispersions
2. First-Principles and Empirical Approaches to Predicting In Vitro Dissolution for Pharmaceutical Formulation and Process Development and for Product Release Testing
3.developing solid oral dosage forms pharmaceutical theory and practice
4.生物药剂学与药物动力学,梁文权著作。
5. Strategies to Address Low Drug Solubility in Discovery and Development
6. The developability classification system: Application of biopharmaceutics concepts to formulation development
<END>

















 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论