



























一、强制降解试验定义
强制降解试验也称为强力试验,是系统稳定性研究的一部分,即根据原料药或制剂产品的性质及剂型特点,通过人为设置较为强烈的条件(如高温、酸、碱、氧化、光照或高湿等)使原料药或制剂产品发生一定程度(5%~20%)的降解。
二、强制降解试验开展的作用与意义
(1)强制降解试验是建立稳定性指示的分析方法的不可或缺的重要手段,通过强制降解试验可以快速获得几乎所有潜在杂质,配合峰纯度考察,可以最大限度的确保方法专属性,避免后期方法因专属性造成的变更及再验证。
(2)在研发初期进行强制降解试验有助于确定潜在的降解产物,并进一步了解产品的降解途径和分子内在稳定性,可以为设计符合监管要求的稳定性计划提供重要支撑同时还可以为包装、贮藏条件以及处方工艺的选择提供支持。
三、降解条件设置
强制降解条件主要包括高温、光照、酸/碱水解、氧化对于易吸湿的样品还应考虑高湿的影响。
表1 强制降解条件类型、原理及参数设置
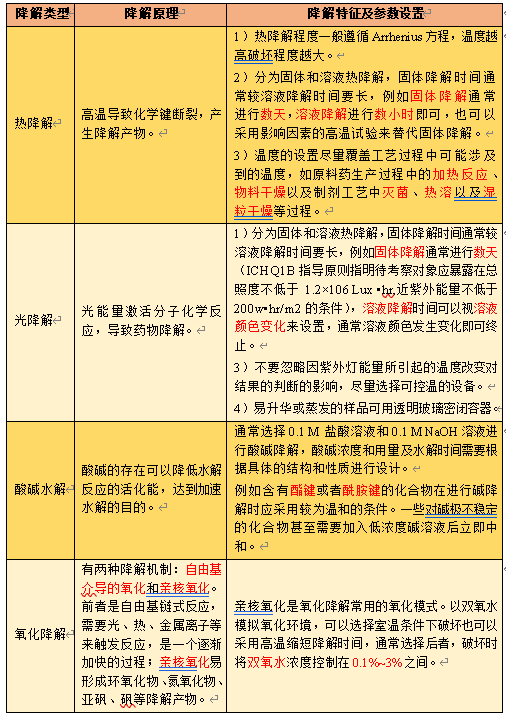
强制降解试验的终点是使样品在个降解条件下均有一定程度的降解,降解杂质得到充分暴露。对于全新的化合物,降解条件不是一蹴而就的,需要先做预降解对化合物在不同条件下的稳定性有大致的判断,然后再根据预降解结果对降解条件进行调整,最后在方法学验证开始之前开展正式强制降解试验。
四、强制降解试验应考察项目
一般在强制降解试验中应考察主峰及杂质与相邻组分的分离度、考察主峰及杂质的峰纯度、计算质量守恒并做初步杂质谱研究。强制降解试验的试验报告中应包含下表的结果。
表2 强制降解试验考察项目汇总
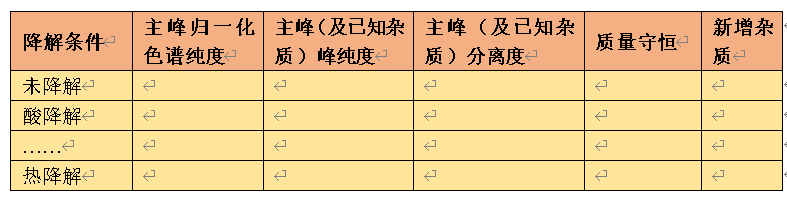
分离度:分离度是专属性的体现,降解杂质与已知杂质及主峰应基线分离,分离度至少1.5。
峰纯度:峰纯度是专属性的补充考察,主要降解杂质及主峰的峰纯度应符合要求。
质量守恒:质量守恒是建立稳定性指示分析方法的重要指标,一般认为质量守恒在90%~110%之间认为是对有关物质评估无显著影响的,误差主要来源于仪器、操作、校正因子、积分设置等。
新增杂质统计及杂质谱分析:应统计各降解条件下的主要新增杂质,必要时采用液质联用法确定杂质分子量,并通过结构特征推测降解杂质的结构。下图(图谱来源于文献5)是替格瑞洛强制降解试验的杂质谱研究结果。

五、强制降解试验结果的提示作用

六、强制降解试验的常见问题
降解程度不足怎么办?
强制降解试验要求主成分降解5%~20%之间,但是在实际的操作过程中某些化合物在较为剧烈的条件下仍无法满足降解需求,这时候其实没有必要再继续增加条件的苛刻性了,因为降解的目的是想了解潜在的降解途径,用于指导稳定性放置条件的设计或者分析方法的开发,对于在剧烈条件下仍不会产生的杂质可以不作为考察对象了。
降解后质量不守恒怎么办?

七、小结
在新药研发过程中合理设计强制降解试验是QbD理念具体实施的一部分,强制降解试验不仅是确保分析方法专属性的手段,还可以为合成工艺选择和处方开发提供依据。
所以,强制降解试验不是照葫芦画瓢或套用固定经验模式的机械劳动,而是需要质量研究人员精心设计,并最大限度挖掘在其中所能获得的关于稳定性及降解机制的信息,助力合成、制剂等学科工作顺利进展。
参考文献:
[1] FDA Perspectives: Scientific Considerations of Forced Degradation Studies in ANDA Submissions,2012,36(5),73-80
[2] ICH Q1A /Q1B
[3] 国家食品药品监督管理总局.化学药物(原料药和制剂)稳定 性研究技术指导原则。
[4] 浅谈化学药物强制降解试验的设计与开展,中国新药杂质,2019,28(20)
[5] 基于AQbD 理念的替格瑞洛原料药杂质研究与质量控制,P29
<END>
要解锁更多企业药品研发信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药品各国上市情况、药品批文信息、销售情况与各维度分析、市场竞争格局、一致性评价情况、集采中标情况、药企申报审批信息、最新动态与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!











 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论