































引言
80%的HCC(乙肝病毒相关的肝细胞癌)是由慢性乙肝病毒(HBV)感染引起,而由HBV基因组编码的乙型肝炎X蛋白(HBx)则是引发癌症的主要原因。作为一种多方面的反式激活蛋白,HBx可以刺激各种原癌基因的表达,包括c-Fos、c-Myc和c-Jun,从而参与促进肝细胞增殖、抑制DNA损伤反应和细胞凋亡的重要致癌过程,并诱导蛋白质降解和HBV复制 ,并且HBx对于启动和维持HBVcccDNA的转录至关重要。
HBx的降解依赖泛素-蛋白酶体系统(UPS),但最近福建医科大学陈婉南团队的研究发现,UPS中DUB(去泛素化酶)也可以对HBx进行调节。相关文章发表在Journal of Virology上,揭示了一种新的HBx调控机制,通过VCPIP1(含有valosin的蛋白质相互作用蛋白1)。

VCPIP1是新型HBx结合蛋白
研究人员通过对74-DUB库进行酵母双杂交试验,发现MPN结构域蛋白(MPND)、泛素羧基末端水解酶22(USP22)、COP9信号体复合物亚基6(COPS6)和VCPIP1 可能与HBx相互作用,针对VCPIP1与HBx进行免疫共沉淀对照试验和反向Co-IP试验,发现VCPIP1与HBx沉淀明显高于对照组。这表明VCPIP1可能是新型的HBx结合蛋白。
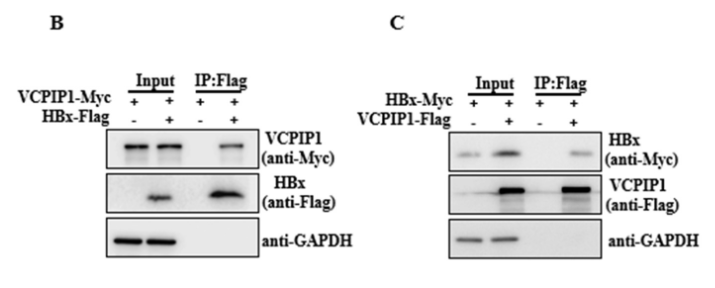
VCPIP1通过延长HBx半衰期增加HBx的表达
研究发现,无论有或者无HBV,VCPIP1都会增加HBx的表达。并且VCPIP1过表达导致Huh7和HepG2细胞中HBx以剂量依赖性方式显著增加。
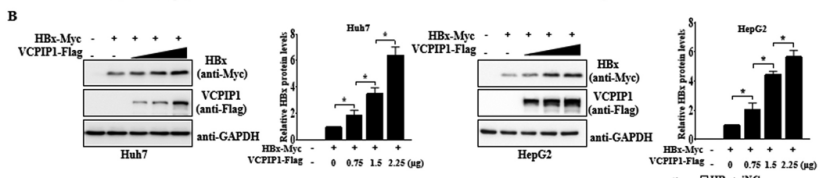
而使用靶向VCPIP1的小干扰RNA(siRNA)(siVCPIP1#1和#2)处理,可以使HBx表达水平显著降低。

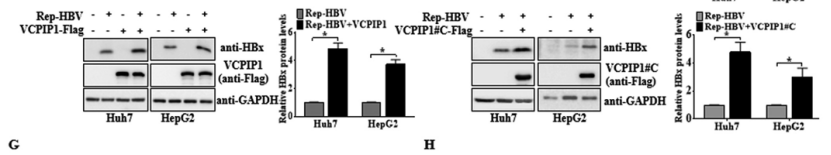
并且在使用含有1.2倍野生型HBV全基因组长度的pRep-HBV质粒感染Huh7和HepG2细胞后,VCPIP1依然能使HBx的表达增加。
而这种表达增加主要是通过延长HBx半衰期的形式实现。使用放线菌酮(CHX)来阻断蛋白质合成,发现,无论是在全长VCPIP1蛋白或aa863至1221相互作用域上,VCPIP1的过表达都将HBx蛋白的半衰期从30分钟延长到了60分钟。
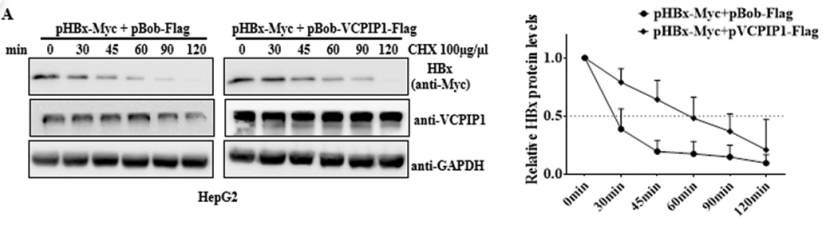
全长VCPIP1-Flag
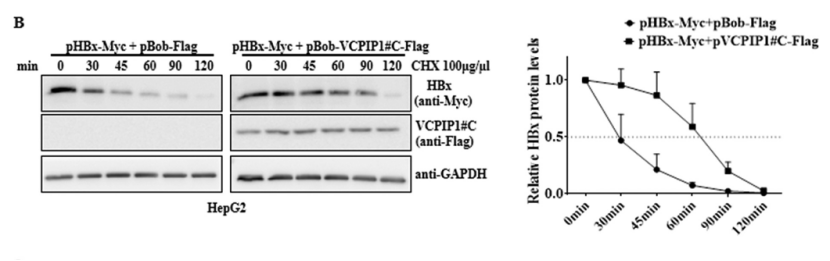
C-末端VCPIP1-Flag(aa863至1221)
相反,抑制VCPIP1导致HBx的半衰期从30分钟减少到20分钟。

VCPIP1通过招募PSMC3,抑制HBx降解,并且是非泛素依赖的
细胞内蛋白质的降解主要由26S蛋白酶体介导,该蛋白酶体由两个19S调节复合物和一个20S蛋白水解核心蛋白酶体组成,此前的作者的研究发现20S蛋白酶体亚基PSMA7介导HBx降解。
考虑到VCPIP1上调HBx的表达 ,作者假设VCPIP1也有类似的机理。对26S蛋白酶的几个亚基(PSMA1、PSMA3、PSMA7、PSMC1和PSMC3 )进行测试,发现在VCPIP1过表达的情况下,仅有PSMC3与HBx结合富集。体外测试PSMC3与HBx结合也抑制了HBx的降解,细胞内测试发现在VCPIP1过表达时异位,PSMC3表达诱导的HBx持续增加。这表明VCPIP1通过招募SMC3与HBx结合形成三元复合物,而抑制20S蛋白酶体对HBx的降解。
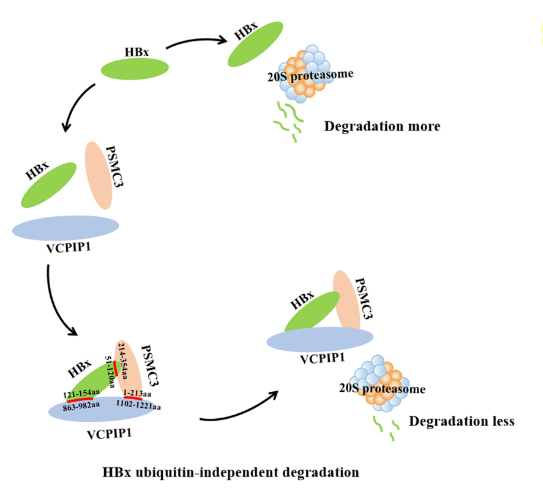
并且这种结合是非泛素依赖的。在体内进行泛素化测定,通过使用标记抗体缀合的琼脂糖珠来沉淀HBx,检测HBx蛋白的泛素化,发现VCPIP1过表达导致Huh7和HepG2细胞中的HBx显著增加,但并没有减少HBx泛素化的量。

而通过泛素化能力受损的pHBxKOR-Flag(HBxKOR(泛素化位点)突变HBx,半衰期与野生型一样为30分钟)质粒试验,发现VCPIP1过表达仍然促进了缺乏泛素化的HBx突变体的表达。
小结
吴琼等人的研究揭示了一种新的HBx降解机制,VCPIP1募集PSMC3可以上调HBx水平。VCPIP1-HBx相互作用促进HBx典型的反式激活活性和细胞增殖抑制。此外,VCPIP1募集PSMC3,提高HBx蛋白水平,并增强HBV cccDNA转录。这表明,靶向VCPIP1或者PSMC3的药物或方法可能能够增加HBx降解,产生更好的拮抗HBV cccDNA的功效,给HCC新药的研发开辟了新的方向。
参考来源:
[1] Hepatitis B Virus X Protein Is Stabilized by the Deubiquitinating Enzyme VCPIP1 in a Ubiquitin-Independent Manner by Recruiting the 26S Proteasome Subunit PSMC3
<END>

















 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论