
































仿制药的历史可追溯到上世纪20年代,最早被仿制的药品是拜耳的阿司匹林。上世40年代晚期,美国已经有仿制药替代的概念出现,但受到当时美国品牌药行业组织“国家药品委员会”(NPC)的激烈反对。他们认为仿制药替代是扼杀创新,而且凭当时的技术理论难以证明仿制药与原研药的一致性。在NPC的作用下,美国医学会(AMA)和美国药师协会(APhA)也站出来反对,他们认为仿制药替代会削弱医生的作用,会违反药品行业的道德和标准。在几大行业协会的影响下,美国的各州开始陆续立法以反对仿制药替代,到50年代末,美国有44个州通过了反替代法。
尽管是反替代,但并不意味着美国没有仿制药,文献数据显示,美国1958年的仿制药处方量占10%左右,不过那时的仿制药都具有商品名,也就是所谓品牌仿制药(brand-generic drug)。60年代以后,美国的医疗支出开始高涨,政府开始重新审视仿制药替代的意义。1962年,美国通过了Kefauver-Harris修正案,为仿制药的替代做了前期的铺垫。根据该修正案,市场上的仿制药被FDA严格监管,要求新药申请(NDA)在获批之前必须完成安全有效性评估,并且要求对1938-1962年间批准的药品开展安全有效性的再评价。
该修正案的实施意味着美国已经开始统一标准,为仿制药替代打下了基础。经过了近10年的再评价,FDA成功地将已上市的仿制药按“有效”、“无效”和“需进一步研究”的标准进行了筛分。1970年,FDA专门为仿制药开通了审评路径,生物等效性试验也成为评估仿制药与对照药一致性的标准。70年代末期,FDA又开始尝试允许一些安全有效性熟知的、1962年以后批准的药品的仿制药申报时使用文献新药申请(paper new drug application),允许仿制药的安全有效性数据引用文献数据。1979-1983年间,有19个仿制药通过这种路径获得了批准。
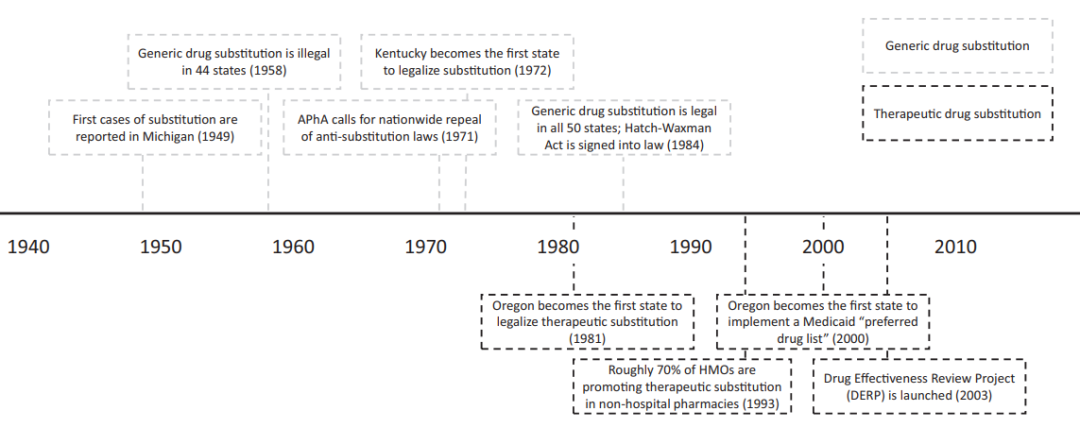
美国仿制药法规体系演变过程的时间轴
虽然FDA在仿制药替代上做了大胆的尝试,但并不意味着监管就此放开。美国是一个法规体系较为复杂的国家,一方面,FDA的各种尝试并未上升为联邦法律,另一方面,各州都有自己的立法权,反替代法并未废除。70年代的美国,已经迎来了创新药发展的黄金时期,大力推动仿制药替代必然会动到创新药企业的奶酪,如何推动仿制药替代,又不伤害创新药企业的利益是当时美国持续争论的话题。好在当时日益高涨的医疗支出开始让政府逐渐向仿制药替代偏移,新生的“药品福利管理组织”(PBM)在其中也起到推波助澜的作用,而且早期反对仿制药替代的APhA也在1971年站出来号召各州废止反替代法。1972年,肯塔基州率先迈出了第一步,随后各州也开始陆续废止该法案,到1984年,反替代法在美国范围内实现了全面废止。
六十年代中期以后,创新药研发逐渐步入黄金时期,大量的新分子实体被开发上市,而这些产品在七十年代末八十年代初陆续失去了专利保护,在巨大的商业前景面前,很多资本家都开始按捺不住。然而1983年时,美国仅有35%的畅销药存在仿制药竞争,而且仿制药的市场份额仅有5%。而且就在同一年,Bolar公司因为提前研发罗氏的地西泮(专利1984年到期)而受到起诉,Bolar公司因被判侵权而成为时代的焦点。1984年秋,Hatch-Waxman法案终获得通过,FDA批准仿制药终有了法律依据,与此同时,反替代法在各州也被悉数废除,仿制药的全面替代终于铺平了道路。根据该法案,仿制药只需要提供简化新药申请(ANDA),通过生物等效性试验和文献数据来代替安全有效性试验,开发成本和周期大幅降低。与此同时,该法案还规定了“Bolar例外条款”,允许仿制药厂家在原研专利期内开展研发、申报和生物等效性试验,鼓励仿制药厂家挑战原研的专利,对首仿药给予180天的市场独占期。因为Waxman-Hatch法案,美国仿制药的巨大潜力瞬间被释放,FDA统计数据显示,1984年,美国有150个专利到期的品牌药没有仿制药,而就在第二年,FDA收到了1050项新仿制药申请。
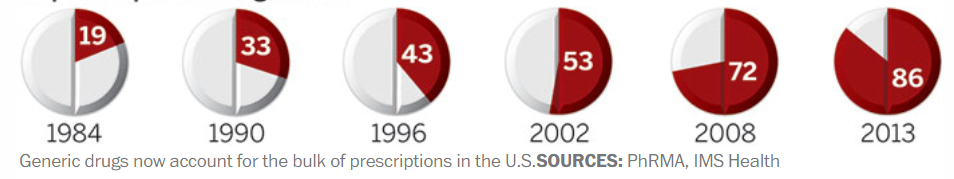
美国仿制药的处方量变化
Hatch-Waxman法案为仿制药开辟了仿制药新天地,但并没有扼杀创新,有效地平衡了“仿制”与“创新”的利益关系,也推动了创新药的发展。根据该法案,创新药可得到不超过5年的专利期限补偿(批准后总有效专利期不超过14年),以弥补创新药在研发和申报过程中的专利时限损耗。一方面,创新药企业的利益得到有效的保障,创新的积极性得以大幅提高,另一方面专利悬崖成了他们悬在头顶的“达摩克利斯之剑”,倒逼他们持续不断地创新来实现企业的基业长青。
因为Hatch-Waxman法案,仿制药的准入门槛被大幅降低,而仿制药也因此不再是稀缺的资源,在一定程度上更像是短生命周期的“快消品”。因为Hatch-Waxman法案赋予了首仿药市场独占期,而抢占市场独占期就是美国仿制药盈利的主要方式。在市场独占期之后,仿制药的价格随着竞争厂家的增多而迅速下降,到最后甚至无利可图。基于这种特点,仿制药企业必须不断地申报新ANDA来维持企业的生存,在自行申报不足以满足的情况下,就只能通过兼并和收购。
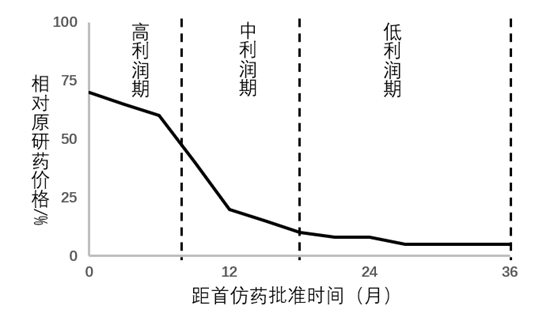
仿制药的生命周期
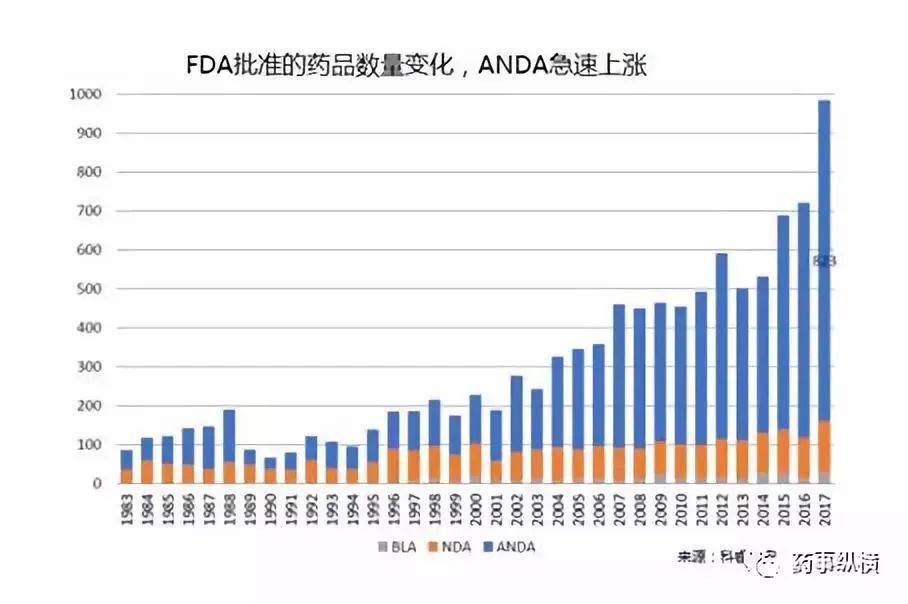
自Hatch-Waxman实施以来,美国一直在坚定不移地推行仿制药替代政策,先后出台了多项配套措施鼓励仿制药申报。随着ANDA数量的不断增加,美国的仿制药竞争开始白热化,数据显示,FDA每年批准的ANDA数量从80年代中期的50个左右迅速增长到2002年的200多个,如今已近1000个,而美国制药处方量也从1984年的19%迅速上升至2002年的52%,如今已超90%。因为竞争导致价格持续下滑,2016年之后,美国仿制药市场呈现出萎缩的态势。为了增加仿制药的申报积极性,近年以来,FDA又开通了仿制药竞争疗法(CGT)的新审评通道,鼓励开发竞争不充分的产品,并对授予CGT资格的仿制药给予180天市场独占期。除此以外,FDA还在探索统一仿制药评估标准,寄希望于申报资料在ICH范围内互认,以降低申请人的开发成本,增加仿制药的申报。
因为美国政府不能直接干预药品价格,所以美国并没有国家层面上的集采,但在GPO(药品集中采购组织)和PBM的多方作用下,仿制药因为残酷的市场竞争,价格几乎达到全球最低水平,90%以上处方量的仿制药仅消耗了美国药品总支出的18%,2020年为美国节省药品支出高达3384亿美元。
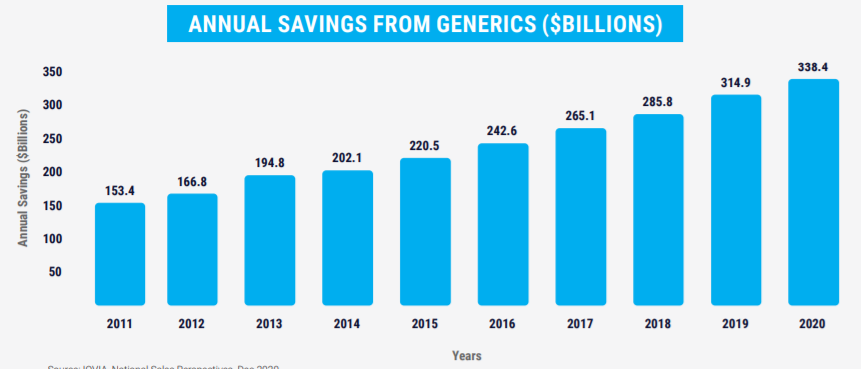
仿制药每年为美国节省的医疗开支(AAM)
Hatch-Waxman法案是一部影响深远的法案,以至于其精髓被全球广泛借鉴。渐渐地,品牌仿制药逐渐退出了历史的舞台,而标准统一的仿制药替代成为国际大趋势。在美国之后,欧洲、加拿大、日本等国也积极探索了特色化的仿制药替代。注册审评方面,这些国家与美国大同小异,在准入和支付政策上,欧洲率先实施了集采,日本则采用了谈价的政策。尽管这些国家采用的方式有所不同,但基本是殊途同归,德国、英国、西班牙、加拿大、法国和日本等国家在仿制药替代上都取得了极大的成功,而这些国家也都是全球屈指可数的大市场,这些国家和美国的仿制药价格逐年下滑带动了全球仿制药价格的下降。
中国虽然没有Hatch-Waxman法案,但美国法规体系的精髓已经被借鉴。从“临床自查”开始,到“注册制度改革”、“一致性评价”、“动态GMP实施”、“上市许可人制度”、“加入ICH”,再到“专利链接制度”,我国的申报和监管法规体系正在向国际靠拢,仿制药价值回归、与国际接轨是必然的趋势。2018年之后,我国又引入了“国家集中带量采购”、“地方性带量采购”、GPO采购等多种招采模式,原研药的“专利悬崖”效应和仿制药替代效应逐步快速地显现了出来,与此同时,仿制药的价格也呈现出剧烈的下滑。
随着集采和一致性评价的深度推进,国际仿制药市场的诸多特征也在我国市场上逐步显现出来,而我国仿制药企业的运作方式也必须快速与国际接轨。在这种背景之下,要么使用国际仿制药巨头惯用的方式(挑战专利抢首仿、构建技术或商业壁垒)来赚取相对高附加值的回报,要么利用信息的不对等性布局特色品种以获得短期的主导定价权,否则只能靠拼规模、拼效能、拼成本来赚取“塞牙缝”的利润。然而,时至今日,还有很多人没有放弃品牌仿制药时代“大品种”、“大市场”、靠一两个仿制药平步青云或“一招鲜吃遍天”的幻想,这种幻想不但会影响企业的产品布局,而且还会让企业遗误转型的良机。
尽管仿制药的利润正在因集采变得越来越鸡肋,但在降价的同时,还应看到高速增长的处方量需求。我国目前人均处方量仅有美欧日等发达国家的五分之一到三分之一,未来增量巨大。另外,笔者一直认为集采导致的“过度降价”是暂时的,一方面是我国的集采政策还处于调整和完善期,另一方面是最早实施集采的欧洲已经在思考如何有效保证各供应链环节的利益以维持产业的持续性发展。因此,在销量和销售额的“剪刀叉”之下如何平衡利润是仿制药企业需要重点思考的问题。在笔者看来,任何时代都不会背离“产品为王”行业法则。不论是国际仿制药巨头,还是中国的中小型仿制药企业,批文数量越多,机会就越多,抗压能力也就越强。因此,仿制药企业不仅要积极申报新ANDA批文,而且在必要的情况下还要积极报团取暖。
总之,我国仿制药仍大有可为,只是在布局仿制药之时,须看清趋势,顺应形势。品牌仿制药是缺医少药时代的特定历史产物,仿制药不会再有大品种,大市场,更不会再有一招鲜吃遍天……
参考文献:略
以上内容仅代表作者个人的见解,欢迎广大朋友批评指正,但未经作者本人同意,严禁任何媒体抄袭转载,侵权必究!
<END>

















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论