































随着医保政策与集采步伐的加快,药企在不断更新的医药环境中均趋于创新,一方面为人类健康贡献力量,另一方面可以稳定自身的市场占有率。然而随着IND和NDA递交的增长,给医药研发人员也带来了一定的困难与挑战,如何迅速从自己仿制药的前认知中抽离出来,很快的置身于创新药的知识体系构建中来。依然可以从问题的本质出发,即药品的安全性、有效性和质量可控性。分析人员首当其冲的着眼于质量可控性,药物研发中的质量可控性在于制定的标准可以有效的控制药物的质量,在整个质量标准中被关注更多的当属杂质研究。所以,本人结合研发经验浅谈一下新药中非无机的杂质方法形成过程,希望与广大同仁共同探讨。
一、列出信息清单
采用逆向思维将拟定方法需要的信息清单列出来。
举例:拟定采用HPLC-UV的方法进行杂质方法开发与建立,需要的信息清单如下:
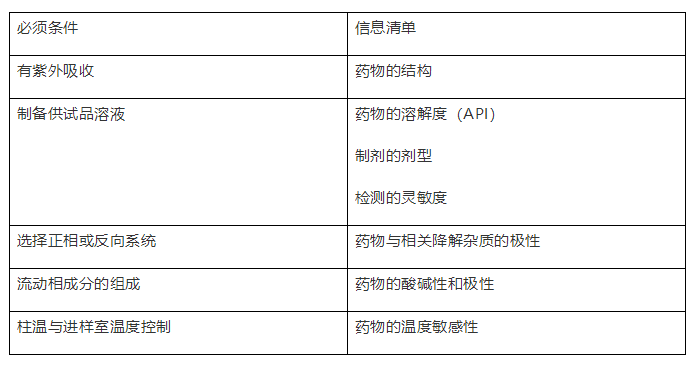
二、杂质谱分析
拟建立的分析方法的根本要素是对杂质的检出能力,因此,对目标药物的杂质谱分析至关重要。采用正相思维层层分析杂质存在情况。
举例:起始物料经过4步合成后得到目标药物,目标药物、辅料及注射用水制成灭菌的注射液,拟分析目标药物的杂质谱。
1、原料药的杂质谱
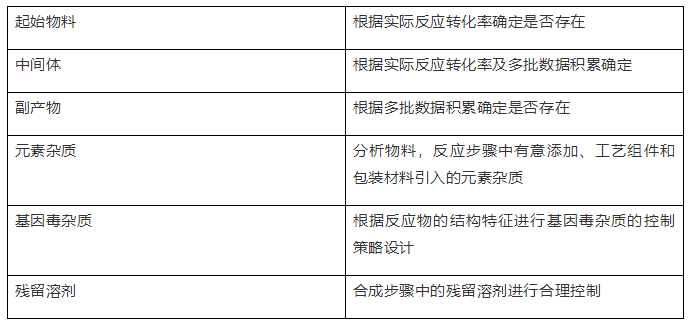
2、制剂的杂质谱
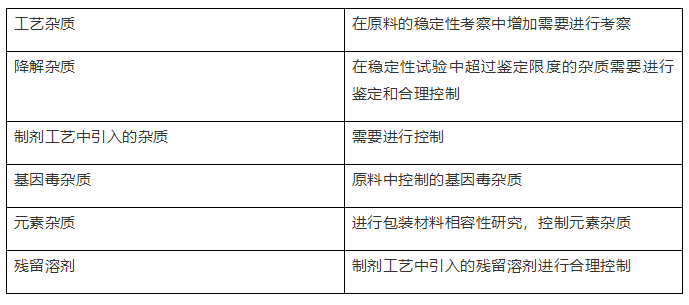
三、可接受标准的拟定
杂质检查的目的是控制特定杂质不超过特定的限度,未知单个最大的杂质不超过一定的限度,总杂不超过一定的限度。可接受的标准如何拟定是控制新药质量的关键点。
举例:新药注射剂的最大日服剂量为600mg,分析各杂质可接受标准的方法。
1、基因毒杂质的限度计算
如果属于1类致突变杂质,根据特定的PDE值,结合每日服用的最大剂量,可以得出该杂质的限度PDE/最大日剂量。
如果属于2类致突变杂质,一般根据TTC值进行控制,即1.5μg/天,结合每日服用的最大剂量,可以得出该杂质的限度AI/最大日剂量。
2、元素杂质的限度计算
根据每种元素的PDE值,结合结合每日服用的最大剂量,可以得出该杂质的限度PDE/最大日剂量。
3、特定的有机杂质的限度计算
该杂质作为特定的有机杂质说明其超过了ICH规定鉴定限度0.2%,需要结合该杂质的毒理学数据如TD50,进行十万分之一的线性外推,得出特定杂质的PDE值,然后结合每日服用的最大剂量,可以得出该杂质的限度PDE/最大日剂量。
4、最大未知单个杂质的限度计算
参考ICHQ3A和Q3B中的杂质限度要求,可以用鉴定限度来控制最大未知单个杂质的限度。
5、总杂的限度计算
总杂的限度制定一般需要结合稳定性的数据,最后进行确定。
6、残留溶剂的限度计算
参考ICHQ3D中的杂质限度要求进行合理控制。
四、杂质分析方法的形成
结合以上三部分的分析之后,确定了药物的杂质谱,限度信息,同时对于HPLC-UV的方法所需的清单信息进行研究,可以初步的制定出供试品的制备方法,对色谱系统进行选择,即确定好色谱柱,根据药物的结构选择流动相,此处流动相的筛选结合药物的结构特征,针对性的查找已有标准的化合物的流动相系统,同时也可以根据药物的溶解性,选择相应的洗脱溶剂。关于流动相系统的选择稍微展开一下:
举例:API溶解于甲醇中,可以应用甲醇制备API的供试品溶液,以C18色谱柱开发方法,流动相选择甲醇-水的系统,起始可以采用1:1的流动相配比,如果出现色谱峰形大三角等异常,可以考虑增加缓冲盐优化峰形,至于梯度方法结合待分析物合理优化。
另外,需要考虑的是基因毒杂质,一般杂质的限度很低,普通的HPLC方法难以满足检测的灵敏度,因此,可以基于基因毒杂质小于限度的30%的方式,在中间体中进行控制,因为在中间体控制中乘以清除率,可以提高杂质的限度,对于仪器的灵敏度的要求就没有那么苛刻了。
综上所述,IND与NDA中的杂质分析方法并非无迹可寻,分析人员只要确定好思路一样可以较为快速的制定出IND与NDA中的杂质分析方法,推动项目的有序进展。
参考文献:
ICHQ3A:新原料药中的杂质
ICHQ3B:新制剂中的杂质
ICHQ3C:杂质:残留溶剂的指导原则
ICHQ3D:元素杂质指导原则
ICHM7:评估和控制药物中DNA反应性(致突变)杂质以限制潜在致癌风险
<END>


















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论