


























“授人以鱼,不如授之以渔”,可谓流传千古,奉为圭臬。但你是否想过,可能只是自己的一厢情愿?可能只是自己好为人师而已?这个问题当然不只是存在于药学人之中,这是一个社会性问题。
以笔者从业领域来说,具备了相应专业能力的前辈通常愿意“授之以渔”,但往往碰到的都是要“鱼”之人。细观慎思之后发现,要鱼之人对于“渔”,并不是不愿意追求,只是想追求更高效、更简单的方式。比如,炸鱼:丢。砰!捡。药鱼:洒。漂!捞。
看出上面“渔技”的特点了吗?对,就是渔死鱼!我们可以反思一下,自己是否也是这样的渔夫?你说我不愿意学习吗?那我一般是不愿意承认的!因为炸鱼、要鱼这种简单高效的方式我的确学会了!而且能解决大部分的鱼,甚至渔获已经富足奔小康了。您倒是复杂,又是下钩,又是埋网,可惜常常寒江独钓!
注意!本文从该行开始,追求走上一篇略显专业文章的道路,上面那是扯扯!由于笔者涉足药学研究的“分析”部门,下面抛“鱼”引“渔”,妄论几段,以供批判!
关于化合物强制降解实验
因为有啰嗦和好为人师的毛病,所以上鱼之前还是想叨叨两句。这个话题我相信很多人查阅过,请教过,讨论过,只要一提,肯定能够各抒己见、指指点点。你没看错,就是指指点点。最后指点结论:具体问题具体分析!不得不说,干的漂亮,真是标准答案,比CDE还CDE!
多数人可能会认可的是,如果化合物很容易降解,这个实验难点就在于降解产物的分析,包括鉴定、分析方法的建立等,恨不能只要降解出来一点就要鉴定,就要建立针对性的分析方法吧?反之,如果不容易降解,这个实验的纠结之处就在于实验条件的终点如何确定,总不能为了满足所谓的破坏量,无限制的去破坏吧?
重要提示:强制降解实验是探索性实验,不是验证性实验。
本文主要说仿制药:首先查阅文献,一定要站在巨人的肩膀上。但前提是,你得能爬上去,站稳了。
降解量一般控制在5-10%,如果降解产物比较纯净(出现1~2个或几个特征杂质峰,未伴随“杂草丛生”),降解量可以更大。
废话不再叨,上鱼(更多针对原料药,制剂可借鉴)!不过这是条大鱼,小店确实没能力上一整条,先上几块,吃着可以的话,找其他地方再踅摸去吧。
第一块:高温。
温度敏感型:室温破坏不同时间,见好就收!
温度不敏感型:条件好选,但破坏到啥时候是个头?
实验室允许高温设备连续运行,100℃放置5天,不同时间点检测;否则,可选择120℃放置9个小时,当天结束。期间可设置不同时间点取样检测,见好就收,不见好也收!
第二块:高湿。
湿度敏感型:现成的稳定性实验箱(40°C ± 2°C/75% RH ±5% RH)安排上,见好就收!
湿度不敏感型:可选择120℃回流9个小时,见好就收,不见好也收!
第三块:光照。
光敏感型:按ICH条件放置,见好就收!
光不敏感型:按ICH条件放置30天,设置不同时间点检测,见好就收,不见好也收!
第四块:氧化。
敏感型:室温,双氧水浓度探索一下,见好就收!
不敏感型:30%双氧水室温放置10天,设置不同时间点检测,见好就收,不见好也收!
第五块:酸、碱。
敏感型:室温,酸、碱浓度探索一下,见好就收!
不敏感型:1M的浓度,60℃放置10天,设置不同时间点检测,见好就收,不见好也收!
另外可以参考的思路是,咨询一下合成实验室的同事,原料药结构形成的那一步反应条件以及后续精制步骤的实验条件,如果涉及到温度、酸、碱、氧化等,重新设计并加剧那些条件直接进行一轮破坏实验,这个结果也是有很好的参考意义的。
最后一大块:分离鉴定!
有些化合物在有些破坏条件下,降解的相对单一,路径很明确,比如酯基的水解等。如果碰到这种情况,那分离鉴定当然就不是问题了:直接把主成分一降到底,顺手拿个杂质对照品。
说到把主成分一降到底,此处插一个小问题。我们经常看到一个论点:降解量不能过大,否则容易出现二次降解,这是没有必要的。不过这是真的吗?其实这是值得商榷的,至少不是一句放之四海而皆准的话。比如说,谁能保证主成分降解的某个杂质一定老老实实待在那儿一动不动等着你去检测呢?它不会变个身?
举个可能有点代表性的例子:奥硝唑注射液的制作工艺过程中,发现在高温灭菌后会产生一个降解杂质A,但杂质A可没那么老实,慢慢就成了杂质B,这算不算二次降解?
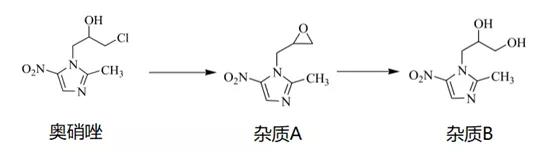
图1 奥硝唑降解路径
上面所碰到的情况,那都属于前世修来的福气,如果破坏完发现了那“杂草丛生”的一堆色谱峰,如何面对?比如下图2所示的降解图谱,虽然降解杂质峰分离的还不错。
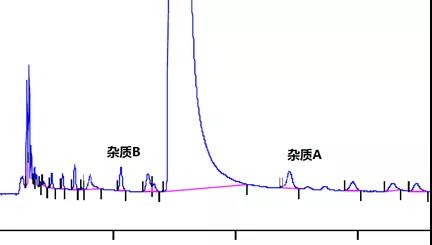
图2 某原料药被某条件强制降解后的HPLC-UV图谱
上图2中某原料药总破坏量在5%~6%,除了已知的两个杂质(杂质A与杂质B)外,还有不少无名氏出现,那需要对它们一一指名道姓吗?还是说挑一些优秀的认识一下?挑谁呢?古语有云,天塌下来个高的顶着。所以,可以考虑挑个高的!
具体怎么挑?可以参考下面的表格及附属的两个例子:
来源于辉瑞全球研发实验室出品文献:Therole of degradant profiling in active pharmaceutical ingredients and drugproducts(Doi:10.1016/j.addr.2006.10.006,请原谅没有按科技论文形式做引用)。

例1:假设某原料药破坏实验中降解总量为25%,其中有一个主要的降解杂质占16%,另一个稍大些的占3%,其他杂七杂八加起来不超过2%。
- 占比16%的杂质,代表了降解量的60%以上,是主要降解产物,需要给予分离鉴定
- 占比3%的杂质,虽然超过了降解总量的10%,但不超过最大降解杂质的25%,因此判定为非重要杂质,无需分离鉴定。
- 那些不超过2%的杂质,不超过降解总量10%,同样不超过最大降解产物的25%,因此均可判定为非重要杂质,无需分离鉴定。
例2:某化合物的光照强制降解试验显示产生了10%的降解量,出现了所谓的“杂草丛生”,其中最高一根占比0.8%。
由于该降解试验中无任何单个杂质超过降解总量的10%,因此均无需进一步分离鉴定。
上面例子中所描述的方式可以让我们在实际操作中不知所措时,有一些借鉴,当然,还是那句废话:具体问题具体分析!
鱼已上完,以上几块各位看官暂且品着。离席之前,再送一条小鱼可带回养之赏玩:说起强制降解试验,免不了涉及到分析方法,以及分析方法验证中的一些做法。
前文的重要提示说过,强制降解试验是探索性试验,不是验证性试验,但偏偏在方法学验证的专属性试验时,“要求了”采用强制降解试验证明方法的专属性以及具有稳定性指示作用。
这里的“要求了”是真的强制要求了吗?怎么说的呢?请看中国药典通则9101关于专属性的描述原文:
在杂质对照品可获得的情况下,对于杂质检查,也可向试样中加入一定量的杂质,考察杂质之间能否得到分离。
在杂质或降解产物不能获得的情况下,可将含有杂质或降解产物的试样进行测定,与另一个经验证的方法或药典方法比较结果。也可用强光照射、高温、髙湿、酸(碱)水解或氧化的方法进行强制破坏,以研究可能的降解产物和降解途径对含量测定和杂质测定的影响。
药典应该不会说废话,也不会说没有逻辑的话,这点我们要去相信她!那对于方法验证专属性中强制降解试验应该如何对待?药典其实说的很明确,我们总结一下:
1、如果通过前期的文献调研,以及探索性试验研究,已经获取了各个条件下典型的降解产物对照品,在此基础上建立的有关物质方法,当进入方法学验证时,无需再次进行强制降解试验。
2、如果获取不到某些条件下的降解产物,可以在方法学验证时提供该条件下的降解试验及图谱,以证专属性。
3、只有当任何一个条件下的降解产物都没有分析出来,也没有获取对照品的情况下,才需要在方法验证时一股脑的提供全部的强制降解试验及图谱,以证专属性。
那么当我们真的需要在方法学验证时进行强制降解试验时,该如何把握呢?抛一砖想引玉,希望没有砸到人。
首先,保证主峰纯净。
其次,拟关注的降解峰之间以及与其他工艺杂质峰之间分离度需满足方法可用(不一定是1.5,有可能需要2.5,也可能只需要1.0,最终需要用试验设计来验证)。
此外,如果“杂草丛生”且“不分伯仲”,尝试通过试验设计找出降解时首先出现的降解峰,保证其纯净,此时不必强求5%以上的降解量。
好吧,就抛这么多砖,以下为本文使用说明,重要提示!
有人说,你上面叨叨一大堆,又不是官方文件,照着干废了怎么办?嗯,确实有这个风险,最后真有可能营养没增加,吃的闹肚子。但是官方一般都是给“渔”,哪有给你“鱼”的呢?既然这样,那不如暂且把上面这些鱼拿回去加以烹调,再行食用,而且吃自己爱吃的,其他的该吐就吐掉。切忌生吞!









 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论