
































起始物料(Starting Material)既是原料药注册申请通用技术文件(CTD)中工艺描述的出发点,也是药品生产质量管理规范(Good Manufacturing Practice,GMP)第一次实施的引入点,还是今后药品生命周期管理(Life cycle Management,ICH Q12)的起始点。对于化学合成原料药而言,起始物料的选择是一个首要的问题,也是原料药药学审评的重点及难点。
一、化学起始物料指认的相关法规指南
ICH Q11 (原料药的开发与生产),2012年5月
EMA《原料药化学指南(草案)》,2015年2月
FDA要求行业按照ICH Q11指导原则选择起始物料并阐明理由,2016年2月
CDE发布的《化学药品新注册分类申报资料要求(试行)》(2016年第80号文)要求简述起始物料选择确定的合理性依据,明确规定起始原料的选择应符合ICHQ11及欧盟的相关技术要求,2016年5月
二、起始物料指认的基本原则
基于ICH Q11指导原则和问答文件指南,起始物料指认的基本原则核心要点如下:
a) 起始物料是原料药结构的重要组成单元
b) 起始物料具有明确的化学属性和完善的质量控制标准
c) 起始物料距原料药之间有多步反应
d) 起始物料杂质的影响和控制策略的充分性
e) 多个起始物料的适用性
f) 起始物料供应商应有完善的生产与质量控制体系
起始物料的选择和论证的详细过程应表述在3.2.S.2.3物料控制部分。在论述起始物料的选择时,要采用适当的科学原理进行论述,考虑整个合成方法和控制策略,结合上述所有原则,而非仅严格遵循单个原则。通常,申请人选择少数几个标准对起始物料的选择进行论述,例如,“化合物X特性清楚,被分离,具有清楚的化学特性和结构,成为原料药的重要结构片断,因此根据ICH Q11它被选择作为起始物料”。这样的论述不够全面,因此不会被接受。仅仅只有控制策略的话,是不足以作为起始物料的论证依据的。同样,合成路线很长并不能弥补很差的控制策略。因此上述起始物料指认的原则要一一论述和阐述原由
三、 起始物料指认常见问题
申报资料中常见的关于起始物料指认的问题如下:
(1)原料药首次注册申报时,拟定的工艺路线仅包括一步化学合成步骤;或在原料药批准后的生产工艺变更申请中,拟缩短原工艺路线,但均未进一步论述拟定起始物料的选择依据;
(2)未结合起始物料的合成路线,对起始物料中实际/潜在存在杂质进行相关的质量研究;
(3)未明确起始物料质量标准的拟定依据;
(4)未提供针对起始物料供应商详细的审计报告,未明确是否同供应商签订质量保证协议和变更管理协议;
(5)起始物料结构过于复杂。
四、起始物料指认中ICH M7指南的应用原则
申请人应识别出可能在生产工艺中生成或引入的致突变杂质,同时需要确定哪些步骤产生的致突变杂质达到了影响原料药杂质谱的水平。
ICH M7指南可以用来确定哪些实际存在或潜在的杂质被认为是具有致突变性的。
对于选择和论证起始物料,建议采用以下方法:
a) 应该评估原料药中已经确定的杂质(实际存在的杂质)的致突变性。
b) 应该评估从市售化学品到原料药合成过程中所用的试剂、中间体的致突变性。需要注意的是,最终所选起始物料之前步骤中的一些试剂、中间体的致突性也应该考虑进行评估。
c) 致突变杂质如果是市售化学品或合成中间体中的杂质,或者是合成副产物,也可能会出现在原料药中并达到与安全相关的水平。申请人应使用基于风险的推理来确定在哪些步骤应该对此类潜在杂质进行危害性评估,如果确认了合成过程中哪些步骤需要对这些杂质和副产物进行致突变性评估,则需要进行风险评估讨论。
d) 如果一个致突变杂质在原料药中的含量高于ICH M7规定的可接受摄入量的30%,则通常认为该杂质对原料药杂质谱有影响。在这种情况下,控制策略通常包括对杂质在可接受限度水平的检测(见ICH M7第8部分)。ICH M7所描述的所有方法均可以用来确定哪些杂质可能超过限度的30%。
e) 根据ICH M7和ICH S9,在某些情况下(例如,当原料药本身就具有致突变性,以及这些指南中所描述的其它情形),选择原料药起始物料时并不需要特别考虑上述水平的致突变杂质谱。在这些情形下,如果致突变杂质不超过ICH Q3A规定的鉴定限度,则认为对原料药的杂质谱没有影响。
五、起始物料的论证案例
基于ICH Q11指南,在确定起始物料之前应对化合物的一些重要信息加以理解。申请人应对生产工艺有充分的认识以理解杂质的形成、去向和清除过程,包括理解影响原料药的杂质形成;理解杂质在生产工艺中是如何衍生和清除的;评估任何可能引入的基因毒性杂质杂质的风险;理解控制原料药质量所需要操作条件变化的影响。
在申报资料3.2.S.2.2生产工艺部分应对生产工艺进行充分的描述,充分性包括:首先确定哪些工艺步骤会影响原料药的杂质谱;考虑将需精细控制的直接上游步骤包括在S.2.2部分;如果只有少数的化学转化步骤,那为了减少由于污染或将来工艺路线变更导致的风险,一步或多步工艺需加入S.2.2部分;控制策略在降低变更导致的风险方面的角色应当加以考虑。
5.1关于持续存在杂质的研究案例
在ICH Q11案例4中,杂质1、2、3会影响原料药杂质谱,此时不局限于是手性杂质。杂质1是步骤1中生成的化合物B的手性杂质。该杂质会持续存在于原料药中。原料药中其它的明显杂质产生于步骤4、5 和6中,如图1。杂质2和3分别在步骤4和5中生成,在步骤2和3中没有产生影响原料药杂质谱的杂质,此时选择化合物D可作为起始物料。此时如果涉及致突变杂质,还应在选择起始物料时运用ICH M7的原则进行论述。
如果杂质2产生于步骤3,其它条件不变,则化合物D不再适合作为起始物料,应重新选择指定化合物C为起始物料。
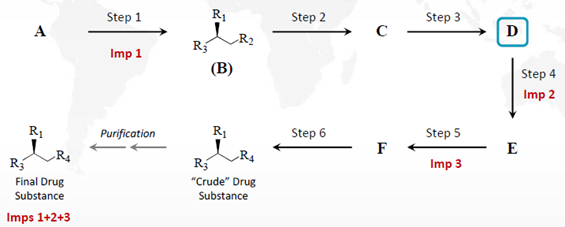
图1:ICH Q11案例4
如果杂质1、2、3分别产生于步骤1、2、3中,则化合物C也不再适合作为起始物料,应重新指定化合物A作为起始物料。
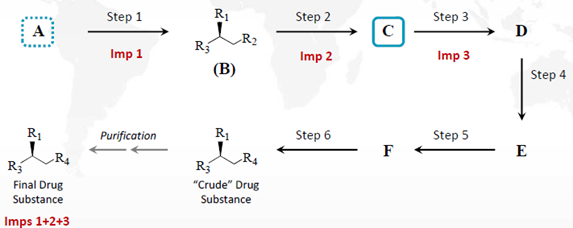
图2:ICH Q11案例4拓展
5.2关于路线过短的研究案例
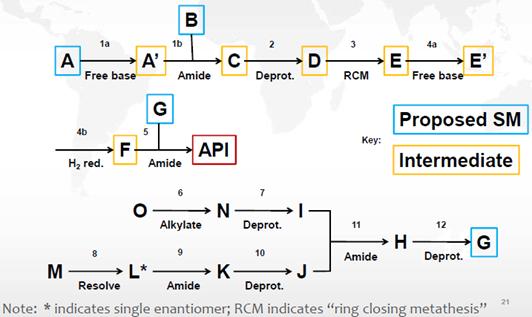
图3: 应用ICH Q11问答指南和决策树的起始物料指认案例
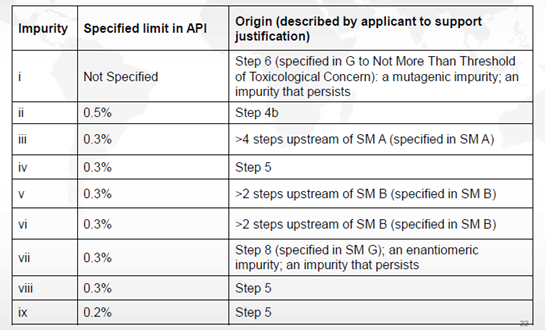
上述合成反应的杂质情况如下表所述
申请人提出将图2中的化合物A、B、G作为起始物料。通过分析其合成路线(图2)、杂质来源,以及运用ICH Q11问答和决策树,结论如下:
a) 步骤6和8中产生的杂质会持续存在于原料药中。杂质i产生于步骤6中,为持续存在的致突变杂质。
b) 化合物I和J之前的步骤不产生其它影响原料药杂质谱的杂质,S2.2中不需要包括步骤6-10。
c) 通过ICH Q11问答指南和决策树分析,化合物A、B、G符合下述的要求,但对于化合物G仅有一步的化学转化步骤列入S.2.2步骤中,因此化合物G不适合作为起始物料。
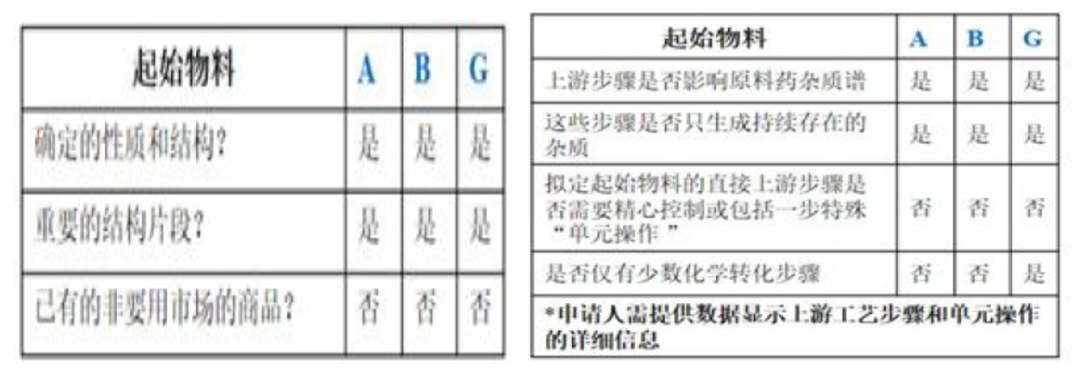
d) 将化合物G作为中间体,同时将化合物I和J作为起始物料进行论证是可以接受的。重新定义起始物料后,就有了充分的工艺描述。
5.3化合物结构复杂和控制策略的研究案例
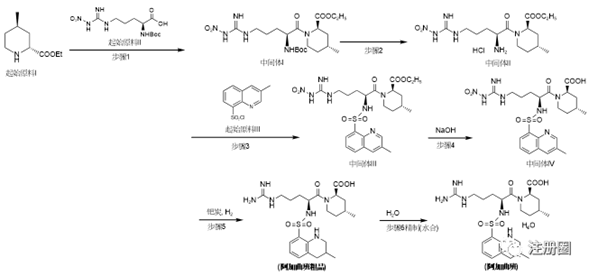
图4:阿加曲班的氨基保护法合成路线
申请人直接以中间体I作为起始原料合成阿加曲班,如图4。中间体I已经具有三个手性中心,属于结构较复杂的化合物,没有将其手性中心的引入过程纳入合成工艺,因此,需将起始原料I和起始原料II纳入到注册工艺中,并从源头起始原料I和起始原料II对其可能引入的异构体杂质及其他工艺杂质进行控制,来保证后续成品的异构体及杂质符合限度要求。
起始原料III通常以3-甲基喹啉盐酸盐为原料,在氯磺酸中高温磺化得到3-甲基喹啉-8-磺酸,3-甲基喹啉-8-磺酸经氯化亚砜氯化后,加入冰水后离心得粗品,粗品经乙酸乙酯重结晶后得产物3-甲基喹啉-8-磺酰氯。
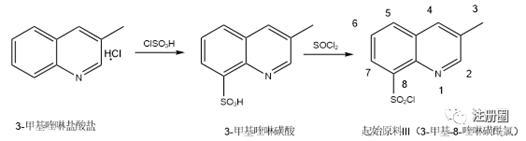
图5:起始物料III的制备工艺
根据起始原料III制备工艺及反应机理分析,磺化反应为喹啉环的芳香亲电取代反应,故主要的产物为3-甲基喹啉-8-磺酰氯。由于苯环与吡啶环共轭体系的影响,即使存在异构体,喹啉环的5位、6位、7位很难发生取代,通过购买或制备其他位置异构体进行检测,多批次中均未检出,故不在起始原料III中进行控制。此外,对制备起始原料III的原料3-甲基喹啉盐酸盐及中间体3-甲基喹啉-8-磺酸在起始原料III中进行研究并做相应控制。反应过程的氯化亚砜为警示结构杂质,根据ICH M7的控制方法4,由于其极不稳定,后续有精制及水洗的过程,不可能残留于成品中,故不进行控制。起始原料III的质量控制得到了很好的控制,适合作为起始物料。
六、总结
案例1中的杂质1作为持续存在的杂质进行了论述。杂质2和3作为非持续存在的杂质,由于影响了原料药的杂质谱,因此也就影响到了起始物料的指认步骤。
案例2中化合物A和B符合起始物料决策树的所有条件,因此可作为起始物料;化合物G由于仅距原料药一步的路线,对于原料药的质量控制有高风险,因此化合物G不适合作为起始物料,合成G的前两步反应物料I和J更适合作为起始物料,对于原料药的质量风险影响较低。
案例3主要从化合物的结构复杂度方面排除了中间体I作为起始物料,而选择了结构更简单的起始原料I和起始原料II作为起始物料。起始物原料III由于其质量可控,其异构体杂质和基因毒性杂质都可以很好的得到控制,不影响原料药杂质谱,因此适合被指认为起始物料。
化学合成反应中起始物料的指认和其质量研究是一个系统工作,需根据ICH Q11中提到的全部原则一一进行论述。起始物料的质量应有足够的控制,除含有持续存在的杂质处,应不对原料药杂质谱产生影响,是其被指认的必要条件。
作者简介:曾文亮,从事新药研究与注册工作,个人公众号:“文亮频道”
参考文献:
ICH, Development and manufacture of drug substances(chemical entities and biotechnological/biological entities)Q11.
ICH, Development and manufacture of drug substances(chemical entities and biotechnological/biological entities)Q11 Q&A
化学合成原料药起始物料的选择原则,王云等,中国医药工业杂质,2022
探讨化学合成原料药中起始原料的相关要求,龚青等,中国药事,2019
阿加曲班原料药发补控制策略分析,注册圈,2022
<END>




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论