



























引言
在开始注射剂项目研发生产前,系统的处方前研究必不可少,详细而全面的处方前研究会让大家少走很多弯路,如(1)参比制剂停产或退市了,(2)项目进行到一半才知道要做临床试验,(3)原辅料投错了量,等等。
今天,咱们就一起来通过(1)药品基本情况,(2)参比制剂/国内上市制剂/原辅料基本情况,(3)注册分类、参比制剂及是否需要临床研究这三方面来系统的了解下处方前研究。
注射剂项目研发生产策略全解析是以仿制药举例论述的,创新药在诸多方面亦可参考。
一、药品基本概况
1、国内外研发历史
1.1国外研发历史
YYDS是三菱化学公司和神户大学冈本浩助教授于1978年合成的世界上第一种选择性抗凝血酶药物,在1990 年1月,用于“慢性动脉闭塞《伯格氏病/阻塞性动脉粥样硬化》”这一重要性适应症批准后被重视,于1990 年6月由日本田边三菱株式会社研发上市了YYDS注射液,后续于2000年获美国食品药品管理局(FDA)批准上市, 2001年获韩国和加拿大批准上市,2002年获中国批准进口上市。
2005年3月,在YYDS注射液(XXml:XXmg)基础上,日本田边三菱株式会社进行小规格的配方开发,成功研发了(Xml:Xmg)规格,该规格处方比原处方具有更高的浓度,于2005 年 7 月,以YYDS注射液(Xml:Xmg)获批上市。随后于2006 年3月,日本田边三菱株式会社YYDS注射液(XXml:XXmg)规格非质量原因停产,停产原因不详。
1.2国内研发历史
天津药物研究院药业有限责任公司于2005年5月在国内率先上市YYDS注射液(XXml:XXmg),博瑞制药(苏州)有限公司于2022年3月在国内上市YYDS注射液(Xml:Xmg),随后国内多家制药企业开启了仿制追逐之路。
2、销售情况(统计至少近5年的数据)
(1)全球销售情况;
(2)国内医院销售情况;
(3)社会面销售情况(非处方药重点关注);
(4)各公司销售占比;
3、重点关注
查询YYDS注射液(XXml:XXmg)及(Xml:Xmg)国内外研发历史,是为了了解该品种的研发上市历史沿革,重点关注原研制剂出现停产、退市等情况,一定要深究其根本原因,避免因为原研制剂由于质量原因导致的停产、退市使自身的仿制进度中断,这样可以提前避免这种风险的出现。
查询YYDS注射液(Xml:Xmg)国内研发进度及销售占比情况,根据自身研发进度及该产品线在市场的占有率,提前推算出该品种未来的销售情况,确定是否立项该品种。
4、资料收集途径
1、https://db.yaozh.com/(药智数据库,很全面,90%以上的资料都有)
2、https://xueshu.baidu.com/(百度学术,查制剂文献资料等)
3、https://www.baidu.com/(百度,查上市历史、销售数据等)
4、https://www.drugfuture.com/pmda/(日本上市药品数据库,查处方、说明书等)
5、drugfuture 药物在线-快捷药物信息平台-DrugFuture数据在线-药物数据
6、FDA橙皮书www.accessdata.fda.gov/(查询FDA认可的参比制剂信息)
7、EMA https://www.ema.europa.eu(查询欧盟集中审评的药品资料)
8、HMA www.drugfuture.com/hma(查询欧盟成员审评的药品资料)
二、参比制剂/国内上市制剂/原辅料基本情况
1、参比制剂


2、国内外上市进度
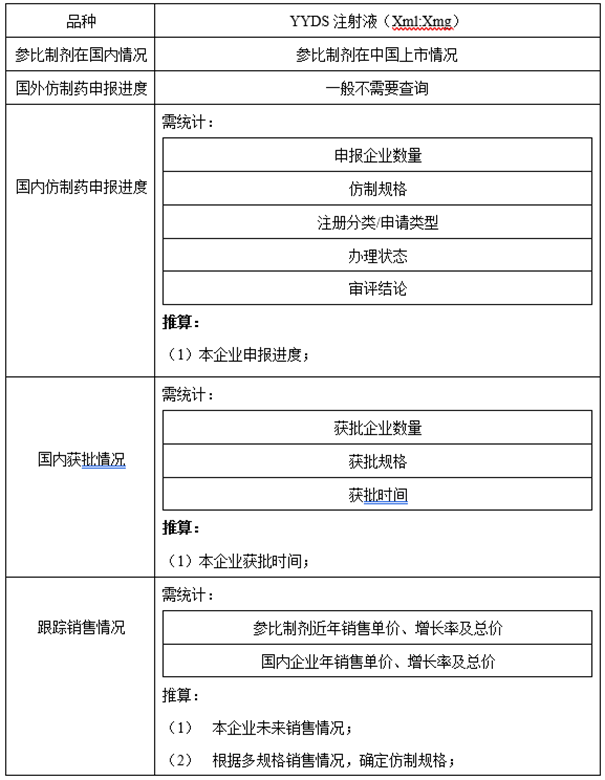

3、资料收集途径
1、https://db.yaozh.com/(药智数据库,很全面,90%以上的资料都有)
2、https://xueshu.baidu.com/(百度学术,查制剂的文献资料等)
3、https://www.baidu.com/(百度,查上市历史、销售数据等)
4、https://www.drugfuture.com/pmda/(日本上市药品数据库,查处方、说明书等)
5、https://db.ouryao.com/(中国药典、法规及标准,查药品及检测标准等)
6、https://www.cde.org.cn/(国家药品监督管理局药品审评中心,查原辅料上市信息及指导原则等)
7、https://www.canbigou.com/directory.html(参比购,查参比制剂目录)
8、https://www.drugfuture.com/fda/(美国FDA药品数据库(U.S. FDA Drugs Database),查处方、说明书等)
9、https://www.drugfuture.com/ema/(欧盟EMA药品数据库,查处方、说明书等)
10、https://drugx.cn/(药研导航,查各国药监局及药典等)
三、注册分类、参比制剂及是否需要临床研究
1、注册分类
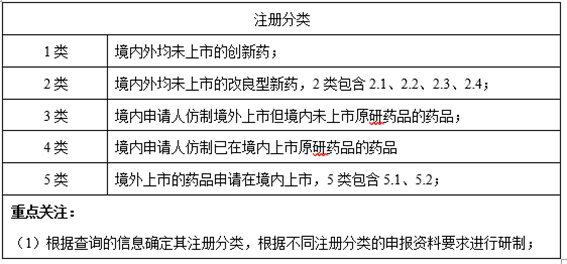
2、原研制剂及参比制剂

3、是否需要临床研究

4、资料收集途径
1、https://wbca.cde.org.cn/wbca/ (参比制剂备案平台,备案参比制剂);
2、http://www.chinadrugtrials.org.cn/index.html(药物临床试验登记与信息公示平台,查询临床试验登记情况);
3、https://www.cde.org.cn/zdyz/index(国家药品监督管理局药品审评中心,查指导原则,关注最新注册分类及临床试验要求);
4、《化学药品新注册分类申报资料要求(试行)》(关注注册分类及临床试验要求);
5、https://db.yaozh.com/(药智数据库,查说明书,关注生物利用度);
四、总结

第二篇生产线适用性
引言
2021年11月,NMPA发补《药品共线生产质量管理指南》在行业内引起了广泛的关注,其中最重要的思路是相对于传统方法设定的限度(千分之一最低日剂量、10ppm、半数致死量LD50)来说,PDE值在评估清洁残留数据时更具科学性和优势,改变了大家传统的认知。
《药品共线生产质量管理指南》中规定了四类风险需要专用设施或设备进行生产,所以,在项目研发前进行生产线适用性考察是必不可少的。
一、共线生产法规
1、强制专线情况
《药品共线生产质量管理指南》中规定了持有人和药品生产企业应当按照法律法规要求,综合考虑药品的特性、生产过程、预定用途、厂房设施与设备等因素,评估多产品共线生产的可行性,并形成共线生产可行性报告。
当药品具有如下风险时,需要专用设施或设备进行生产:
(1)法律法规、国家标准明确使用专用设施或设备的(例如:高致敏药品、性激素类避孕药等);
(2)毒理学评价得出的科学数据不支持交叉污染风险可控的;
(3)毒理学评价得到的相关残留限度不能通过已验证的分析方法检出的;
(4)污染和交叉污染风险不能通过操作过程和/或技术措施得到充分控制的。
2、是否能够共线情况
(1)化药与生物药、化药与中药最好不要共线;
(2)最终灭菌产品和非最终灭菌产品共线,可以共线,需要按照非最终灭菌产品共线进行过程管理;
(3)某些激素类、细胞毒性类、高活性化学药品共线
①生产性激素类避孕药品,必须使用专用设施;
②某些激素类、细胞毒类、高活性化学药品最好阶段生产;
3、根据OEL评估能否共线
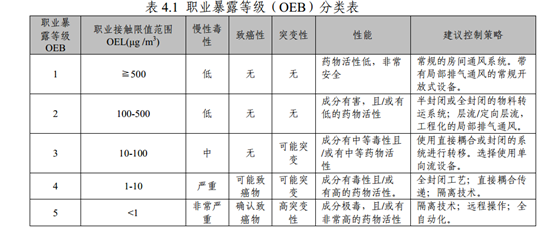
查询参比制剂安全说明书,确定产品OEL,根据产品OEL评估是否能共线。
4、根据ATC分类评估是否能够共线
依据产品ATC分类及上述信息,确定产品是否能共线生产,若不能则改用专线或使用一次性技术进行生产。
二、计算产品的基于健康的暴露限度(HBEL)
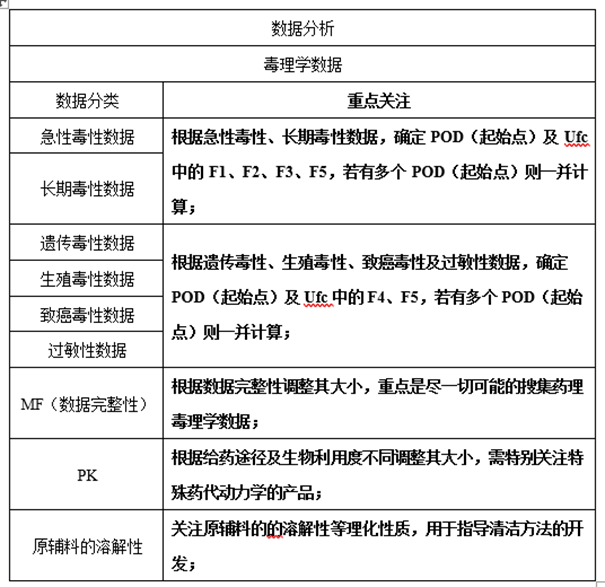
1、毒理数据收集
在药品研发前期,要充分收集产品毒理学的相关数据,如急性毒性试验、长期毒性试验、遗传毒性试验、生殖毒性试验、致癌毒性试验、过敏性试验等的相关研究数据,用于计算基于健康的暴露限度(HBEL)。
收集原辅料的溶解性等理化性质,用于指导清洁方法的开发。
2、HBEL的计算方法


3、数据分析
三、确定生产线
1、自有产线
根据上述信息,确定了不需要专线生产,可以使用自有产线,需考察自有产线相关信息,如:
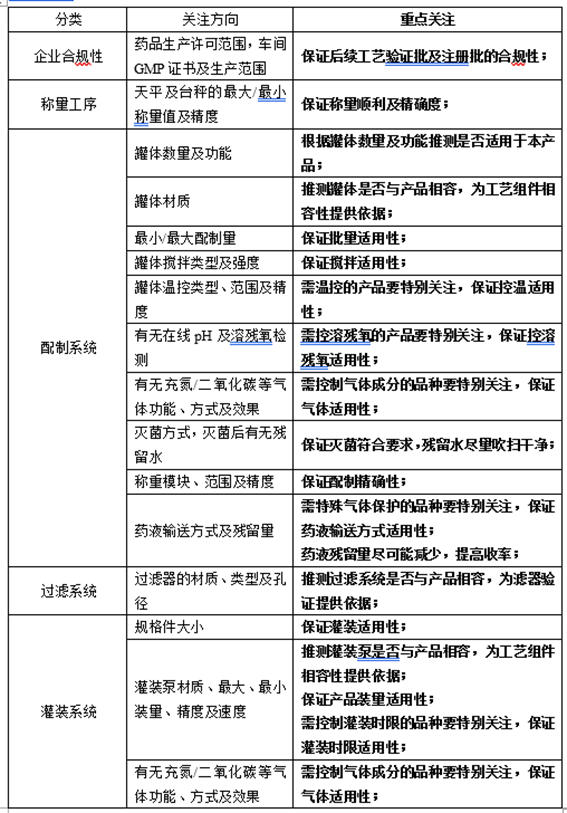
2、委托生产
根据上述信息,确定了不需要专线生产,自己若没有相应产线,则需要考虑委托生产,除考察“1、 自有产线”相关信息外,还需考察,如:(1)该公司仓库物料管理系统,(2)QC检测适用性,(3)员工专业性,(4)资料合规性,等等,更需要熟悉产品的制剂、分析、质保等多部门实地考察。
3、需专用产线
3.1专用产线
根据上述信息,确定了需要专线生产,考察项目同 “1、自有产线”相关信息一致。
3.2一次性使用技术
根据上述信息,确定了需要专线生产,该产品附加值高、批次少,公司从成本出发决定使用一次性使用技术。
一次性使用技术也称可抛弃型技术,是在整个系统中和药品接触部分采用了一次性使用的部件,这些部件在一次或几次的使用后作抛弃处理。
使用一次性使用技术,需要用户方、设计方、供应商多方充分沟通交流,根据实际厂房布局及要求,充分考虑工艺风险,设计出符合需求的方案。
四、总结
根据《药品共线生产质量管理指南》中的规定,科学合理的在项目研发前期考察生产线适用性,是项目顺利推进的基础,
随着共线生产法规的不断更新,基于健康的暴露限度(HBEL)的被广泛认可,一次性使用技术的不断发展,对我们制药工作者,是挑战也是机遇,只有以患者的临床收益为核心,不断提高水平,将挑战转变成机遇,才是吾辈制药人的追求。
相关阅读:
参考文献及资料收集途径:
1、《药品共线生产质量管理指南(征求意见稿)》
2、《关于在共用设施生产不同药品的风险识别中使用的基于健康的暴露限度的指南》
3、《Cross-contamination control and Health Based Exposure Limits (HBEL) Q&As交叉污染控制与基于健康的暴露限(HBEL)问答》
4、ICH Q3C:残留溶剂的指导原则
5、ICH Q3D(R2):元素杂质指导原则
6、ICH M7:评估和控制药物中的DNA活性(致突变)杂质以限制潜在的致癌风险
7、《关于在清洁验证中引入HBEL的考量》
8、《APIC原料药工厂中清洁验证指南》
9、https://db.yaozh.com/(药智数据库,很全面,90%以上的资料都有)
10、https://www.cde.org.cn/(国家药品监督管理局药品审评中心,查最新指导原则等)
11、https://drugx.cn/(药研导航,查各国药监局及药典等)
<END>










 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论