



























新药的发现有一个程序性的步骤,依次深入进行研究,随着研究的深入,标准也依次严格起来,如图1所示。药物发现之初为探索不同的药效团的结构空间,覆盖之广,如大海捞针;然后会缩小筛选的范围,定义先导化合物;在筛选先导化合物的基础上,进行优化,包括构效关系和结构与化合物理化性质的关系。最后进行深入研究,以确定优化后的先导化合物是否适合进一步的开发,成为候选化合物。
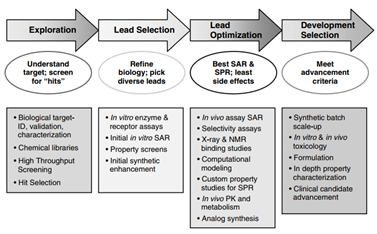
图1新药发现流程图来源于参考文献1
药物发现科学家更加关注化合物与靶蛋白活性位点的结合,以选择强效的候选化合物,往往忽略了化合物的类药性质,例如通过增加化合物的亲脂性提高化合物与靶蛋白的结合,这样造成化合物的溶解度和代谢稳定性降低。化合物强有力的药效不会因为溶解度差而被抛弃,这样就把负担强加到制剂开发人员头上,不得不试图开发高端制剂去解决难溶性问题,例如固体分散体,从以往倾向于开发性质稳定的优势晶型,反其道而行之,开发最不稳定的无定形。这样也增加了制剂开发难度,对于FIC药物或许值得一试,而对于me too或me better,可能会花费大量的时间与资源,推迟上市的时间。
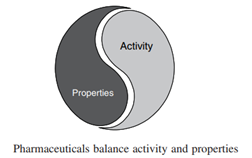
图2药效与药物性质的平衡来源于参考文献2
边缘或者极端的药物的形成来源于药物发现阶段对于药效的一味追求而忽略了药物理化性质的表现,打破了构效关系与结构与理化性质之间的平衡。有些制药公司已经关注到这个问题,一般采取以下措施以减少这种情况的发生。第一种措施为在药物发现阶段早早地进行高通量的动物PK试验,进行化合物体内PK筛选。第二种措施是早早进行化合物体外性质的研究。体外性质研究无需过多的动物试验,将药物的理化性质和化合物的结构联系在了一起。化合物发现科学家可以在早期开发阶段有意识地去兼顾化合物药效和理化性质,如图2所示。化合物的成功开发来自于药效和理化性质或者说类药性的平衡。
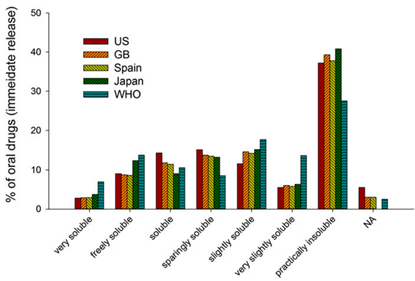
图3 美国 (US)、英国(GB)、西班牙和日本以及世界卫生组织(WHO)基本药物清单中排名前 200 的口服药物产品的溶解度分布比较。极易溶性药物:超过1000 mg/ml;易溶性药物:100-1000 mg/ml;可溶性药物:33-100 mg/ml;微溶药物:10-33mg/ml;微溶性药物:1-10 mg/ml;极微溶性药物:0.1-1mg/ml;几乎不溶的药物:0.1 mg/ml。(来源于2006年参考文献3)
道理如此,现实是新药研发产线中化合物的类药性是十分严峻。尽管试图规避溶解度问题,但目前大约 40% 的上市药物和高达75%的目前正在开发的化合物被认为是水溶性差的,如上图所示。更新的文献报道,正在开发的化合物BSCⅡ和Ⅳ类高达90%,可能现在面对的现实比报道更加严峻。
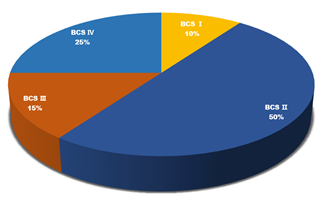
图4 2009年文献报道难溶性药物数据来源于参考文献4

图5 2013年文献报道难溶性药物数据来源于参考文献5
面对难溶性药物的来势汹汹,合适的固态筛选或者可能成为制剂开发人员的救命稻草。一旦确定候选化合物,下一步就要考虑临床前的开发工作,其一就是化合物的固态形式的选择问题。寻找药物开发的最佳形式可能涉及许多不同的筛选活动,包括寻找多晶型物、盐、共晶和/或无定形固体分散体。固体形式筛选活动可以在开发的早期或晚期使用不同的策略在开发的每个阶段进行。无定形固体分散体通常作为该筛选策略的一部分,但它们也可以作为基于溶解度风险评估的制剂策略的一部分,尤其是对于临床前研究。高风险化合物将需要包括无定形固体分散体的特殊配方。无论重点是API还是配方,都可能需要无定形固体分散体,以找到用于进一步开发的最佳形式。
举一个化合物的例子:Telaprevir的难溶性研究
Telaprevir是一种在丙型肝炎病毒中发现的NS3 4a蛋白酶的小分子拟肽抑制剂,蛋白酶对HCV复制至关重要。Telaprevir竞争性地作用于NS3 4a,通过以高亲和力结合蛋白酶的活性位点。仅基于结合亲和力,它作为抑制剂可能表现良好。然而,靶标的性质使事情变得复杂。NS3 4a的活性位点主要是非极性的;因此,只有其他非极性化合物可能与它紧密结合。非极性靶标都是一个难题。非极性配体可能能够与靶位点紧密结合,但可能高度不溶于水或生理相关的水性介质中。Telaprevir具有高度疏水性,因此显示出非常低的水溶性。在水中,它的溶解度低于大理石。由于与靶点作用的需要,Telaprevir所具有的理化性质及生物药剂学性质给制剂开发带来挑战,如图6所示。
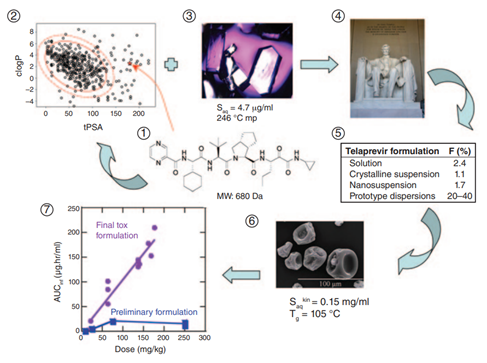
图6 Telaprevir制剂的挑战:(来源于参考文献6)
1.化合物结构导致分子用于口服给药具有挑战性的物理化学特性。
2.高极性表面积(PSA)使得特拉匹韦远远超出了市场上已知吸收良好的口服给药药物的“Egan Egg”之外。需要注意的是,telaprevir测量的logP(辛醇-水分配系数,疏水性的度量)为4.0,显著高于计算2.8(图中所有分子(包括 telaprevir)都使用了 clogP(计算的 logP),为了保持一致性),这使特拉匹韦相对于已上市的口服药物处于logP值的上限。
3.特拉匹韦具有高度结晶性,具有246°C的高熔点(mp),导致结晶材料的水溶性(Saq)极差(4.7μg/ml)。
4.Telaprevir的溶解度实际上低于大理石。
5初步制剂尝试表明溶液、结晶混悬剂甚至纳米混悬剂导致特拉匹韦的口服暴露量非常低(%F是绝对生物利用度)。
6.发现将特拉匹韦配制成无定形喷雾干燥分散体(Tg是玻璃化转变温度)显著提高了在水性介质中的动力学溶解度(Saqkin),从而提高了生物利用度。稳定聚合物和表面活性剂的选择至关重要。
7.毒理学和一期临床研究中使用的早期配方不像最终配方那样物理稳定(即,结晶在水介质中发生得更快)。稳定分散导致显著改善暴露(AUCinf),使Telaprevir的安全范围可以自信地建立。
单独的水溶性既不是有效的口服给药的必要条件,也不是充分条件,但它是开发科学家必须考虑的一个关键参数。由于大多数口服化合物在小肠中被吸收,API必须在肠腔中达到足以驱动其进入体循环的浓度。对于具有较高肠道通透性的化合物,较低的溶解度可能是可以接受的。然而,考虑到它的渗透性,任何化合物都必须具有一定的最小溶解度。
一个化合物的溶解度的过程,包括打破晶格能-形成空穴-化合物的溶剂化,其中晶格能和溶剂化常常作为化合物溶解的限速步骤。晶格能高低与化合物的熔点有关,溶剂化能力的高低与化合物的LogP相关。由上面提供的数据可知,Telaprevir具有高的熔点246°C,晶格能限制了其溶解度。进行合适固态筛选,甚至采用无定型形式或许是一个比较有前途的开发策略。Telaprevir上市产品采用了固体分散体工艺。
阿斯利康在临床前进行固态形式筛选阶段,会在先导化合物或者优化的先导化合物的基础上进行体外和计算机ASD制备的评估,以确定研发产线中的溶解度较差的系列化合物分子是否进行ASD开发。早期选择ASD作为难溶性药物开发的并行策略,不改变化合物结构的情况下,维持化合物构效关系,可以为化合物发现阶段因为难溶性的挑战提供机会(来源于参考文献7)。
总结:化合物固态形式的筛选依旧还是以优势晶型筛选为主(即使筛选盐型或共晶,仍旧需要进一步筛选盐型或者共晶的优势晶型),可是面对难溶性药物的来势汹汹势必需要早做打算,否则开发到最后发现低体内暴露,又需要重新进行固态开发,岂不是造成时间与资源的浪费,如何做好固态开发的顶端设计,将成为制药公司不可或缺的能力,你说呢?
参考文献:
1.Drug-like Properties: Concepts, Structure Designand Methods: from ADME to Toxicity Optimization Advantages of Good Drug-likeProperties
2.Drug-Like Property Concepts in PharmaceuticalDesign (chapter2 Advantagesof Good Drug-like Properties)
3.Strategies to Address Low Drug SolubilityinDiscovery and Development
4.The Innovator Pipeline:Bioavailability Challengesand Advanced Oral Drug Delivery Opportunities
5.A provisionalbiopharmaceutical classification of the top 200 oral drug products in
the United States, Great Britain, Spain, and Japan
6.Discovery and developmentof telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronichepatitis C virus
7.Amorphous solid dispersions: Utilization andchallenges in preclinical drug development within AstraZeneca
<END>











 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论