



























01 摘要
本文介绍了工艺验证生命周期3A阶段的评估方法和相关统计方法。该评估方法可应用于收集了大量工艺和产品数据小分子药物以及生物制品开发。3A阶段可能包括以下要素:3A批次数量的确定;评估关键材料属性(CMA)、关键工艺参数(CPP)、关键质量属性(CQA);体内体外相关性(IVIVC);固有工艺可变性(IPV)和PaCS指数的评估;工艺性能和质量面板(PCQd);加强控制策略。美国FDA关于工艺验证的指南:鼓励将以前可靠经验应用到适当的类似产品和工艺中。对于类似产品和工艺的开发和降低风险,完整的3A阶段评估至关重要,因此需要制定和开发3A评估要素以解决行业和监管指导要求,从而对产品的稳健性做出科学和基于风险的决策。
02 背景
3A阶段是新产品上市后的初步评估,它利用大量数据进行统计评估,以获得更深入的产品理解。3A阶段评估利用来自产品所有工艺验证生命周期阶段的数据(第1阶段:工艺设计;第2阶段:工艺性能确认(PPQ);和第3阶段:持续工艺验证)。2015年7月FDA指南草案:质量指南要求规定:制造控制要素是根据产品上市时对工艺性能的早期估计或使用批准时认为适当的控制策略开发的。在放大和商业制造工艺中获得的知识可用于进一步开发控制策略。3A阶段评估对于理解和管理产品可变性至关重要。
通过评估并密切关注多个确定的批次,以更好地了解可能影响关键质量属性(CQA)的产品和工艺可变性和相互作用(如果有),并通过从基于质量源于设计(QbD)的产品开发活动、技术转移、放大/缩小工艺、设备认证和第3A阶段批次中获得初始工艺资料。本文介绍了Apotex Inc.自2013年4月以来为工艺验证生命周期第3A阶段构思和采用的评估方法和相关统计方法,可以采用表1所示方案和报告。在工艺验证生命周期第2阶段评估完成后,可以启动具有确定要求的第3A阶段方案。完成第3A阶段评估表明企业在建立增强的产品控制策略和获得高水平的产品理解和质量方面的合规性。

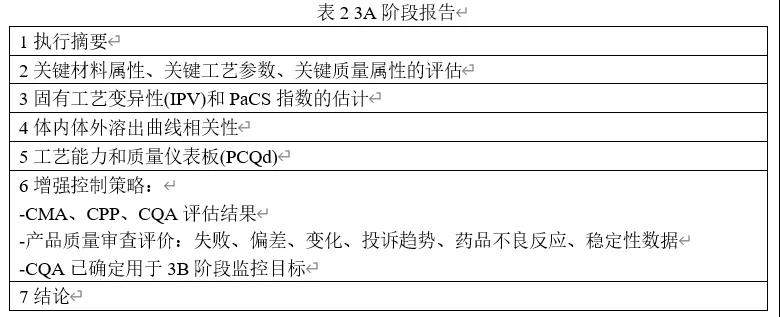
03 确定3A阶段评估所需批次数量
2011年FDA工艺验证指南建议样品数量应足以在批次内和批次之间提供足够的质量统计可信度。Pazhayattil引入了一种新方法来确定工艺性能确定(第2阶段)批次的数量。该方法使用以前收集的产品特定信息和跨多个CQAs的历史批次工艺信息。产品特定信息包括为临床试验、提交或注册、稳定性、工艺放大/缩小和模拟目的而生产的第1阶段批次生成的数据。通过这种方法建立了产品质量属性测量值的置信区间,它是工艺均值的置信区间和工艺标准偏差的置信区间的组合。确定PPQ批次的预计数量,以便整个模拟置信区间位于规格限内。
相同的方法用于确定第3A阶段批次的数量,除了使用第2阶段PPQ批次的结果代替第1阶段的批次内变异性(S0)外。例如,USP<905>剂量均匀性表示接受值(AV)的计算必须小于15才能满足L1标准。现有的批次数据用于确定批次间可变性(SB-B),阶段2数据用于确定批次内可变性(S0)。评估3A阶段批次数量的置信上限为:
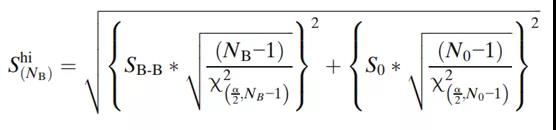
由于每个特定质量属性的测量方式不同,且要求也不同,因此用于确定置信区间的方程是按质量属性制定的。如果根据每个质量属性确定的批次数量不同,建议采用保守的方法选择最高的估计数量。另一种方法可以在样本量计算中使用容差区间,前提是确定所需的批次数,例如,95%的数据将被限制在指定的范围内,置信度为95%。
04 估计固有工艺变异性(IPV)和PACS指数
FDA指出统计工艺控制在理解和管理可变性方面的重要性。控制可变性的主要步骤是了解可变性的主要来源。可变性是由与“6M”相关的各种因素引起的:机器(machine)、人力(manpower)、材料(materials)、检测(measurements)、制造方法(manufacturing methods)和环境因素(mother nature)。在药品制造中,它涉及工艺设备(processing equipment)、人员(personnel)、原材料(raw materials)、分析方法(analytical method)、制造工艺(manufacturing process,)和设施/环境控制(facility/environmental controls)。整体产品(overall product)可变性可以基于估计单个组件可变性来计算,如下面的等式所示:

评估工艺可变性(process variability)的第一步是确保诸如加工设备、人员、原材料和环境控制等可变性影响因素是恒定的。所有3A阶段批次均由经过培训的操作员按照标准操作程序在相同型号的合格设备上进行生产;原材料批次来自同一供应商并符合规格;设施/环境控制和监控系统确保在相同条件下运行。假设这三个因素造成的可变性很小,可以很好的评估来自制造工艺和分析方法的可变性(variability from the manufacturing process and analytical method)。固有工艺可变性(IPV)是批次间可变性的量度,而分析(方法)可变性是批次内可变性的量度。在下面的评估中,抽样可变性和批次工艺中的可变性被假定为最小,并包含在其他两个可变性来源下。如果使用QbD原则开发,则在3A阶段制造的批次预计在批次工艺中具有最小的可变性。

方差分量分析可以通过单向随机效应ANOV 模型进行分析。这种模型通过以下模型方程拟合数据:

其中yij是第i个批次中的第j个用量均匀性(DU)测量值,m是总体平均值,ai是第i个批次对用量均匀性的影响,eij是残差;i=1,…,r和j=1,…,n表示r个批次和每批次n个DU测量值。应该注意的是,在这个模型中,ai被认为是一个随机变量,其中随机条件包括不同的操作人员、设备、批次和天数。
在下面的示例中,整体可变性和方差分量是根据成品用量均匀性(DU)结果估计的。每个3A批次都有剂量均匀性数据,每批次10次测量。每个批次都按照USP进行用量均匀性测试,其中每个单位按照方法进行测试(n=10或n=30)。多批次的DU结果用于计算批次之间(IPV)和批次内(分析)可变性分量。从这个模型中,整体方差可以划分进方差分量,使得:
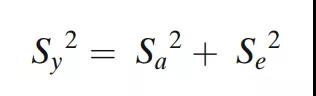
其中Sy2是整体可变性,Sa2是随机变量ai的可变性,Se2是残差项eij的可变性。这里采用ANOVA方差分析估计方法。
假设ai服从均值为0且方差为Sa2的正态分布,并且eij服从均值为0且方差为Se2的正态分布,则Sa2和Se2(并且因此Sy2)的估计值可以推导出如下。
每个批次的平均值表示为yi,总平均值表示为y,r是批次数,n是每批次的结果数。现在让
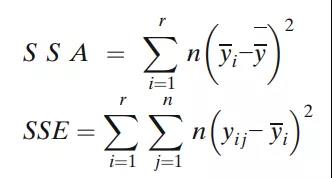
那么,Se2的估计是:
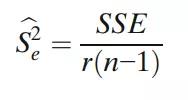
同样,Sa2(IPV)的估计值由下式给出:
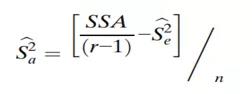
然后将整体可变性作为这两个估计值的总和给出:
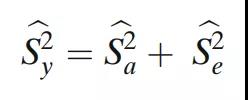
每个组件的95%置信区间(CI)以及整体可变性如下(表3),使用修改后的大样本置信区间。由于工艺引起的方差百分比,即ρBa=Sa2/(Sa2+Se2),分析变异引起的方差百分比,即ρe=Se2/(Sa2+Se2)。因此,此处也包括此类置信区间:
其中F=MSA/MSE;X2c,L和X2c,U是某个水平α的自由度为c的卡方分布的上限和下限,其中Pr{X2c,L≤X2c≤X2c,U}=1-α;FL和FU是某个水平α的{r-1,r(n-1)}自由度F分布的边界,其中;
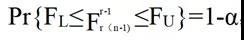

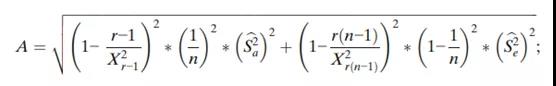
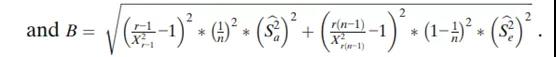
推导上述置信区间的主要假设是模型中的误差项均独立和分布,且均按正态分布。
为了证明这种方法,将单向随机效应模型拟合到产品数据集,以生成方差分量分析。结果在表4中。总体差异为1.84,其中0.97归因于工艺差异,0.87归因于分析差异。因此,就标准偏差而言,可以看到0:971/2=1.00工艺可变性。大约一半的总体方差(53%)归因于工艺变异性(IPV),另一半(47%)归因于分析变异性,如表3中分别在ρa和ρe下所述。表中在估值正下方的括号中提供了95%置信区间。

因此,对于上述示例,固有工艺可变性估计为 0.97。
计算出特定产品的IPV,就可以使用它来推导出PaCS指数。PaCS指数是根据基准衡量的产品性能的计算值。它使用以下公式计算:
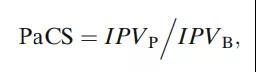
其中IPVB是基准测试固有工艺可变性,IPVp是产品固有工艺可变性。对这些产品中的每一批分别进行方差分量估计。IPVB是与当前产品具有相似工艺的所选产品的中间工艺可变性。与基准相比,PaCS指数>1表示工艺可变性高,PaCS<1表示工艺可变性低。因此,PaCS<1是首选。在PaCS>1的情况下,可能需要进一步评估。例如,如果IPVp大于基准产品的最大IPV,则表明有机会通过持续改进来减少当前产品的工艺可变性。
单向随机效应模型(One-way random effects models)对选定的产品进行拟合,生成一系列方差分量分析,结果见表5。可以看出,工艺方差的范围为0.07(产品9)到1.61(产品1)。正态性和独立性假设通过对每个模型拟合的残差图的适当分析进行测试,并且没有发现严重的违规行为。IPVB,即Sa2的中值,为0.39(如产品7所示)

鉴于上述IPVB为0.39,IPVP为0.97,PaCS指数计算为0.97/0.39=2.5。很明显,该产品的PaCS指数>1,表明工艺可变性高于基准。但是,IPVp小于从所选产品中观察到的最大IPV值(1.61)。如果IPVp大于最大值,则可能需要持续改进策略。
05 体内体外相关性 (IVIVC)
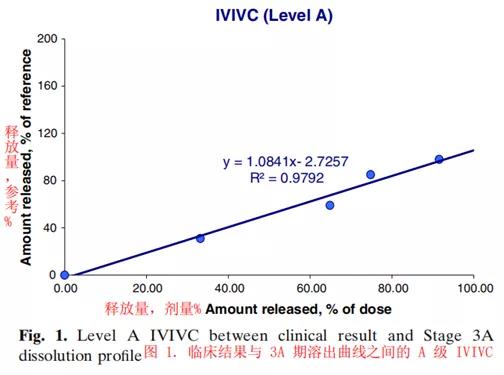
体内生物临床结果与3A阶段体外溶出曲线的相关性进一步证实了产品一致的溶出性能。初始IVIVC是使用中试规模的批次确定的。因此,在第3A阶段相似性评估或IVIVC是有益的,因为它提供保证并进一步支持商业规模生产的药物产品性能,相对于小规模批次。IVIVC源自血浆浓度-时间结果和3A阶段溶出曲线。根据用于建立关系的数据类型,FDA行业指南定义了IVIVC的三个主要级别:缓释口服剂型:体外/体内相关性的开发、评估和应用(Development, Evaluation and Application)。对于信息量最大和最常见的A级IVIVC,相关性是线性和点对点的。体外溶出释放曲线可以直接或通过比例因子与体内吸收血浆浓度-时间曲线叠加。临床结果和3A阶段溶出曲线之间的A级IVIVC是通过确定系数(r2)建立的(图1)。
06 工艺性能和质量面板(PCQd)
产品特定的PCQd是阶段3A评估中预测产品稳健性的关键组成部分。其解决了FDA指南中的要素:质量矩阵请求,其中FDA建议可选指标作为制造稳健性和质量保证的证据。报告的数据表明风险低,可能有助于减少现场检查频率。
每个工艺阶段都可以根据预定的工艺能力目标进行评估,以提供如表6所示的整体产品性能概要。通常专业人员负责选择用于该评估的统计工具,即评估工艺稳定性和能力。PCQd工艺性能目标可以在第3A阶段启动之前根据可接受的工艺性能指标(例如Pp、Ppk)和严格的控制限制设定。
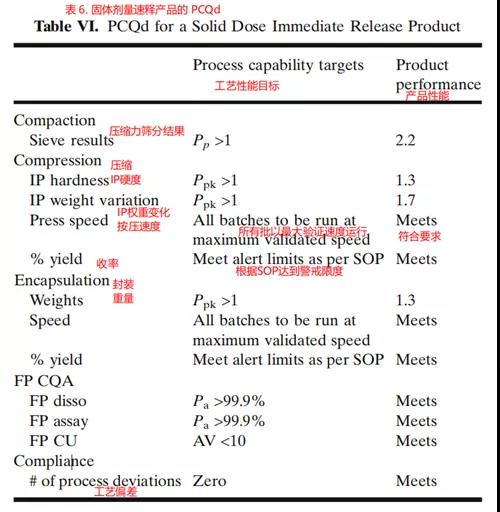
在上面的例子中,Pp公式考虑了标准偏差给出的变异程度和指定限制允许的可接受范围,因此适用于工艺中的CQA,例如筛分分析。对于此IP CQA,样本旨在在整个制造工艺中以预定义的时间间隔满足规范要求。除了工艺变化之外,Ppk评估还能够判断除过程变化外的分散问题。因此,它适用于以满足目标规范为目的的硬度和重量变化。验收概率(Probability of Acceptance,Pa)适用于具有阶段验收标准(例如溶解)的CQA,在这些条件下,传统的工艺能力措施是不够的。
实施PCQd的好处包括:支持主动降低风险的活动,允许通过产品性能监督进行管理层,提高供应链的可预测性和制造可靠性,鼓励实施新兴技术以减少可变性,并使监管机构(例如FDA)能够开发基于风险的现场检查计划。PCQd也可以被组织用作产品开发和负责产品商业生命周期管理的商业运营之间的转移标准。
07 加强控制策略
ICHQ10药品质量体系指南指出,在控制策略中,工艺性能和产品质量监控系统应提供测量和分析明确关键材料属性(CMA)、关键工艺参数(CPP)和关键质量属性(CQA)的工具。基于质量源于设计(QbD)的产品开发数据、鉴定结果和额外的第3A阶段商业批次的结果,最终确定了完善的产品控制策略。ICHQ8(R2)药物开发需要所有关键属性的控制策略。控制策略旨在确保始终如一地生产具有所要求质量的产品。FDA认识到利用可用于增强产品控制策略的上市后研究的重要性。
应该评估用于第3A阶段批次的生产制造的不同批次原材料的CMAs。即使原材料批次符合供应商和内部规格限制,确保在任何已确定的CMAs中产生的变化不会对成品质量属性(如溶解度、含量均匀性和含量)产生不利影响也很重要。药品和/或原料药生产可能需要额外的工艺或规范控制。
在第3A阶段评估的CPPs可以反映任何工艺变化。统计工艺控制图可用于每个CPP来评估工艺参数的可变性。任何生产中发生的变化都可能需要采取适当的行动。CPP是在基于QbD的产品开发的第1阶段定义的。如果观察到CQAs的高可变性,则分析CPPs对CQAs的影响程度,例如,溶解时的预压缩力和主压缩力等参数。可以建立统计模型来理解在进一步评估中可能出现的任何新关系,并作为开发工艺理解和控制策略的关键组成部分。
应该评估每个制造阶段的工艺中质量属性,如压实、压缩和包衣,以及成品质量属性,如分析、溶解和剂量均匀性。尽管CQA存在一定量的可变性,但当它生产的产品随着时间的推移稳定且可预测时,该工艺被确定为处于统计控制状态。统计工艺控制图是工艺监控中最常用的工具。工艺性能分析(Ppk)和验收概率分析(Pa)也用于提供产品性能的量化评估和预测。然后可以使用阶段3A批次的结果分析来证明未来批次满足CQA规范的可能性。
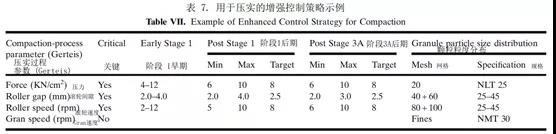
基于风险的科学评估以及实验设计(DoE)研究允许在第1阶段建立控制策略,随后在第2阶段验证并在第3A阶段进一步加强(表7)。定义3B阶段监控计划是产品生命周期管理中增强3A控制策略的一部分。进一步了解变异的来源及其对下游工艺、中间体材料和药品质量的影响,为将控制转移到上游并最大限度地减少对最终产品测试的需求提供机会。满足质量保证的替代方法(例如,用分层剂量均匀性或工艺分析技术,如NIR,代替混合均匀性)在附加数据的可用性下是合理的。
08 结论
本文提供了完成第3A阶段评估所需的方法。已经提到的评估工具可应用于已经收集大量工艺和产品资料的新开发产品和上市小分子以及生物制品。完整的3A阶段评估对具有类似产品和工艺的产品开发和风险减小具有重要作用。3A评估方法的开发是为了满足行业指导要求。该方法系统地评估材料属性、工艺参数、质量特性、固有工艺可变性、药物释放曲线、控制策略和3B阶段的监测标准。项目产品稳健性(如PCQd)和产品可变性(PaCS指数)可用作关键信息,以最大限度地降低供应链风险和确认产品合规性。
<END>









 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论