
































最近,在网上看到一篇文章介绍了“为什么90%临床药物研发失败及如何改进?”。文章解释了药物失败原因主要来源于靶点的验证及药物优化可能过分强调了某些方面,而忽略了其他方面,从而误导了候选药物选择并影响了临床剂量/疗效/毒性平衡,比如造成药物难溶的角度,可以这样解度,强调了疗效,即药物分子作为配体与疏水性受体的亲和力,基于构效关系(structure-activity-relationship,SAP),强化药物分子的疏水,而忽略了药物分子的理化性质,造成分子难溶的局面,破化了结构-性质(structure–property relationships,SPR)之间的和谐。
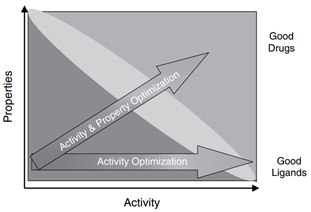
图1 药物活性与性质间的关系(来源:文献1)
造成临床药物研发失败的原因,对于靶点验证的工作确实离我们做制剂的人员来说有点远,针对第二点原因我想制剂人员是可以从制剂的角度进行认领,同时也可以明晰制剂工作如何提高新药开发的成功率或者确认一下新药制剂开发的首要小目标。因为候选药物在人体试验中的成功离不开临床剂量/疗效/毒性的平衡,临床剂量/疗效/毒性的平衡不仅取决于其抑制分子靶标的活性/特异性,而且还取决于其暴露量/选择性在疾病靶器官/组织和正常组织器官中的的平衡(Structure-tissue exposure/selectivity relationship,STR)。一般来说,候选化合物由药物发现部门提供,药物分子与靶点的活性和特异性,药物分子对于靶组织,靶器官VS正常组织器官选择性,这些药物分子的生物药剂学特征由药物分子的结构特点所决定,而在一定程度上,制剂人员通过制剂手段可影响仅仅是药物在体内的暴露量。提到药物体内的暴露量不得不再次和大家说说药物在人体内的ADME过程,如图2模式图所示。
口服固体制剂人体之路
新药常以速释固体制剂(片剂、胶囊)进行开发。一杯250ml水,一粒药,一仰脖,入口腔,经食道,跋涉入胃。胃作为主要食物主要的消化器官,极酸环境,药物一般可以崩解成细小的颗粒,小的颗粒还可以进一步解聚,有些快速或者非常快速释放药物的制剂,在胃部基本已经完全溶出。随着胃排空,“飘洋过海”来到了小肠,小肠包括十二指肠,空肠,回肠。由于其巨大的表面积,血流量高,而且药物停留的时间相对较长,小肠也成为了药物吸收的主要场所。当然,对于碱性药物还是具有挑战的,因由胃入肠,胃肠道环境已经由酸转碱,碱性药物可能由于分子型增多,离子型降低,造成溶解度下降,可能有化合物析出的风险。当然,分子形式药物的增多,提高了药物的亲脂性,给药物穿越肠壁细胞膜提供了机遇。药物一旦透过肠壁细胞后,就可以经肝门静脉进入肝脏。可是,无论肠壁细胞还是肝脏都对化合物可能具有代谢作用,代谢酶对药物分子进行各种各样的修饰,使其失活并被排出。有些药物分子躺尸在了胜利的黎明,成功跨越的药物分子得以继续赶路,幸存的未被肝脏修饰的药物分子才最终抵达心脏,经血液输送到全身。此为药物的吸收之路。
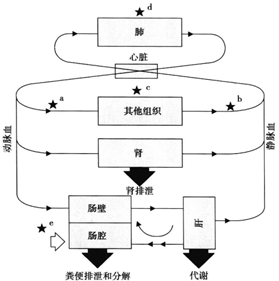
图2 药物制剂的体内过程(来源:文献2)
药物吸收以后,仍然还在路上。药物会在血液循环系统,随血流遍布全身,会扩散进入几乎所有的器官和组织,此为药物分布。药物在不断循环过程中,但凡经过肝脏,都会有一部分被肝脏细胞中的代谢酶修饰,此为药物代谢。最终药物或者修饰过的药物经过胆汁排到肠中随粪便排出,或者经肾脏由尿排出,此为药物的排泄过程。
药物的体内过程在以往所写之文,可能也有所提及,因为制剂开发不仅要关注制剂制备之过程,更需要联系药物制剂在体内之变化。有机地把制剂设计与体内变化联系起来,一来可以根据药物理化性质,加之生理解剖学特征以及生理环境,去预测药物在体内可能具有吸收情况,根据此所设计的预测性工具,也早已经在新药研发过程起到很重要要的作用,例如PBPK模型;二来根据体内的数据可以反馈已经制备的制剂一些情况(如处方工艺),是否需要进行调整,把研发过程,形成一个相互促进的一个闭环,这也是生物药剂学与药物动力学这个学科在药物开发中所具有的重要意义之一。
体内暴露
前文提到了体内暴露这个概念,对于这个耳熟能详的名词一直再提,对于其背后的含义还未有深刻的认识。在此,想试着解释一下。药物在循环系统的带领下,达到靶组织和靶器官,具有一定的药物浓度,停留足够的时间,然后发挥药效,这就是我们常说的药物体内的暴露。暴露量可以是给药剂量,也可以是即时或累积药物浓度,如峰浓度(Cmax)、谷浓度(Cmin)、稳态浓度(Css)和浓度曲线下面积(AUC),最能直接反映暴露量累积情况的是稳态时给药间隔AUC。
在“FDA NDA或IND中生物利用度研究一般考考虑”对于暴露也有一定的阐述。暴露指按照适当生物基质中药物浓度-时间曲线的峰值,局部暴露量和总暴露量来定义。
暴露峰值:即达峰浓度Cmax。达峰时间也可以提供药物吸收的速度Tmax。
总暴露量:包括单剂量给药下的合适的生物基质中浓度-时间从0到t的曲线下面积AUC0-t.;合适的生物基质中浓度-时间从0到无穷大的曲线下面积AUC0-¥以及稳态研究中在合适的生物基质中,稳态时浓度-时间从0到TAU的曲线曲线下面积AUC0-TAU,其中TAU为给药间隔长度。
部分暴露:即部分AUC。
从以上对于体内暴露的描述,简单理解,药物给药后,药物在血液循环中,啥时候浓度最大,最大达到多少,什么时间对应的血药浓度是多少,总的来看有多少药物进入体内。提高体内暴露,就可以简单解释提高药物血药浓度。
通常药物需要有足够的暴露量才能有治疗效果。新药开发早期足够的体内暴露成为追求的一个目标。前文谈到“新药开发早期中最关键需要表征的药物理化性质-溶解度”,通透性也是影响口服药物生物利用度的另一个关键性因素,因此,新药研发的早期阶段,准确及高效的评估药物渗透性至关重要。
- 渗透性
基本概念:渗透性指药物通过某个生物膜屏障的速度,它是药物小肠吸收,通过血液-器官屏障,通透进入含治疗靶标细胞以及肝和肾清除的必要过程。渗透性更多的体现药物跨膜特性,我们常常关注的药物的渗透性更多的是关注药物渗透小肠上皮细胞膜的性质。
- 药物渗透性、吸收生物利用度及体内暴露间的关系
药代动力学是描述药物吸收、分布、代谢、排泄过程的科学。吸收是指药物从给药部位达到体循环的速度与程度。药物的口服生物利用度从数理的角度去描述药物从吸收到入血的过程。药物的口服生物利用度(F)是指机体从药品中吸收API或活性物质并在作用部位产生药效的程度和速度,是药物吸收分数(Fa)、药物排出肠道代谢的分数(Fg)和药物排出肝脏代谢的分数(Fh)的乘积。从方程1可以看到药物吸收决定药物的生物利用度。一个药物吸收由多个过程所决定,包括药物的溶解度和渗透性。

因此,我们可以知道药物渗透性,吸收与生物利用度之间的关系:药物渗透性的增强,可以提高药物吸收入血,药物吸收分数的增加,可以提高生物利用度,药物生物利用度的提高,可提高药物的体内暴露。
当然,对于一个药物分子其渗透性其实难以通过制剂手段去提高的。在胃肠道中,药物分子一旦脱离制剂的制约,药物分子溶解在溶液中。胃肠道的环境,生理解剖特征,药物分子结构及性质,已经不在人为控制之内了,药物分子犹如脱缰之野马,分子型与离子型的比例,主动转运VS被动扩散,是否具有强烈的代谢风险,与血浆蛋白结合紧密如何,对于靶器官、靶组织的选择型如何,这些都与药物自身的性质有关,已经不受制剂所控制。制剂更多的影响药物溶解度和溶出去影响在吸收部门的药物浓度,增加渗透性,去影响生物利用度和药物的体内暴露。(以上说法建立在新药不添加一些吸收促进剂,不考虑一般辅料对药物吸收的影响的前提下)。制剂人员可以不能影响药物分子本身的渗透性作用,但是还是可以通过对于处方工艺的筛选制备最佳的制剂,使药物在吸收部位可以在足够的时间内提供足够的浓度,使药物分子足够的吸收入血,起码做到制剂不会降低药物分子本身的渗透与吸收。。
小结
新药的成功开发,涉及多部门多学科共同的努力,各个环节需要共同发力且缺一不可。新药的开发中,具有很多的不确定性,尽可能提高药物暴露或许成为制剂开发的首要目标。虽然文章想说说渗透性在新药开发的作用,但是这个概念不能脱离药物体内胃肠道这个大环境。渗透性作为可能限制药物吸收又一重要性质,需要把药物吸收、生物利用度和体内暴露联系起来,才能把整个逻辑理顺。当然,本文更多的提到了一些概念,具体如何去表征渗透性,本文还未涉及。近期如何在新药开发中渗透性预测的小文正马不停蹄朝你而来,敬请期待。
新手上路,以上小文难免有考虑不足之处,如有异议地方,还请留言!
推荐阅读:
《新药开发之多晶型理化性质底层逻辑》
参考文献
1 Drug-like Properties Concepts Structure Design and Methods; from ADME to Toxicity(图书)
2. Martin物理药剂学与药学(图书)
3.药物暴露-反应研究的临床设计与评价
4. FDA NDA或IND中生物利用度研究一般考考虑
5.为什么90%临床药物研发失败及如何改进?
<END>




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论