
































导读:近日,CDE上市药品信息栏更新了北京珅诺基医药科技有限公司旗下关于阿可拉定软胶囊(淫羊藿素软胶囊CXZS2101001)的申请上市的技术审评报告。详情见下。
淫羊藿素(阿可拉定)详情
2022年1月,国家药品监督管理局通过优先审评审批程序附条件批准1.2类创新药淫羊藿素(阿可拉定)上市。
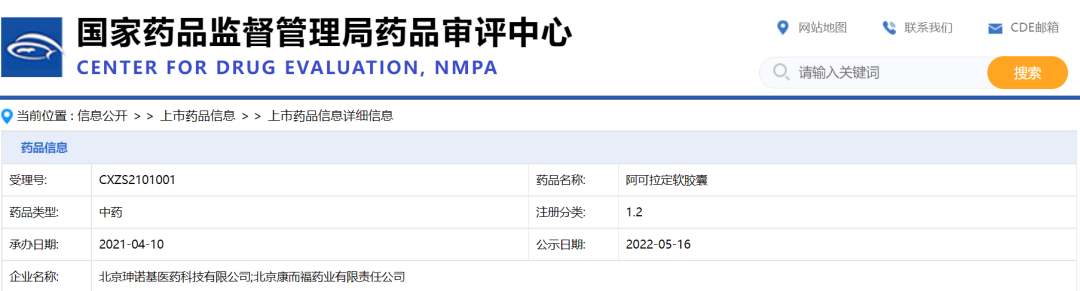
淫羊藿素(阿可拉定)批文信息

截图来源:药融云中国药品批文数据库
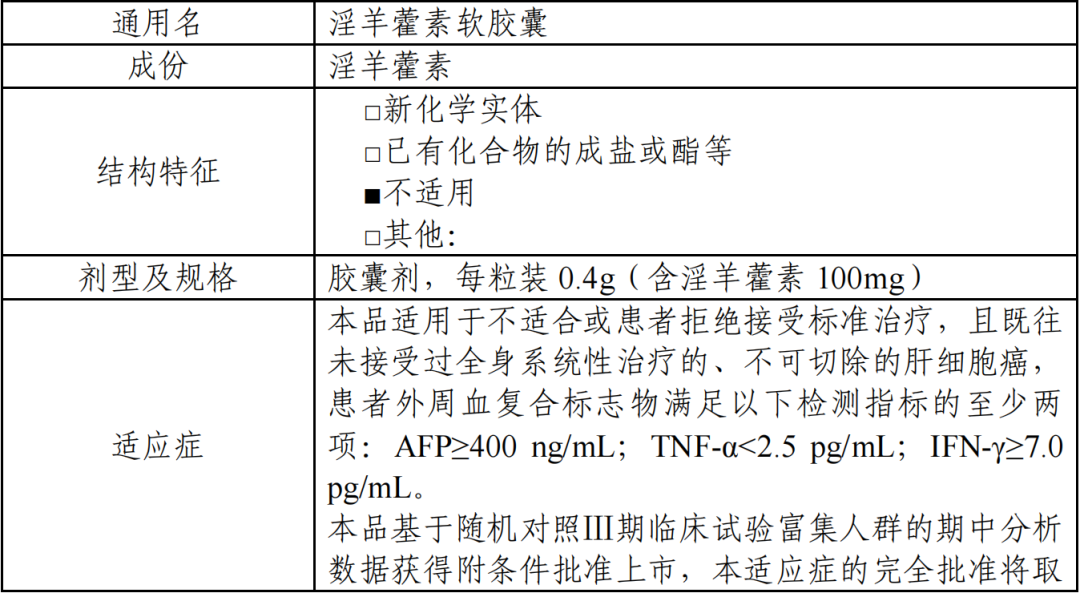
获批适应症:适用于不适合或患者拒绝接受标准治疗、且既往未接受过全身系统性治 疗的、不可切除的肝细胞癌,患者外周血复合标志物满足以下检测指标的至少两项:AFP≥400 ng/mL;TNF-α<2.5 pg/mL;IFN-γ≥7.0 pg/mL。
本品基于随机对照Ⅲ期临床试验富集人群的期中分析数据获得附条件批准上市,本适应症的完全批准将取决于计划开展的确证性试验证实本品的临床获益。
获批规格:每粒装 0.4g(含淫羊藿素 100mg)
另外,据药融云数据库查询,目前申报淫羊藿素(阿可拉定)的企业仅三家,分别是北京珅诺基医药、山东珅诺基医药和北京康而福药业。
淫羊藿素(阿可拉定)CDE承办情况
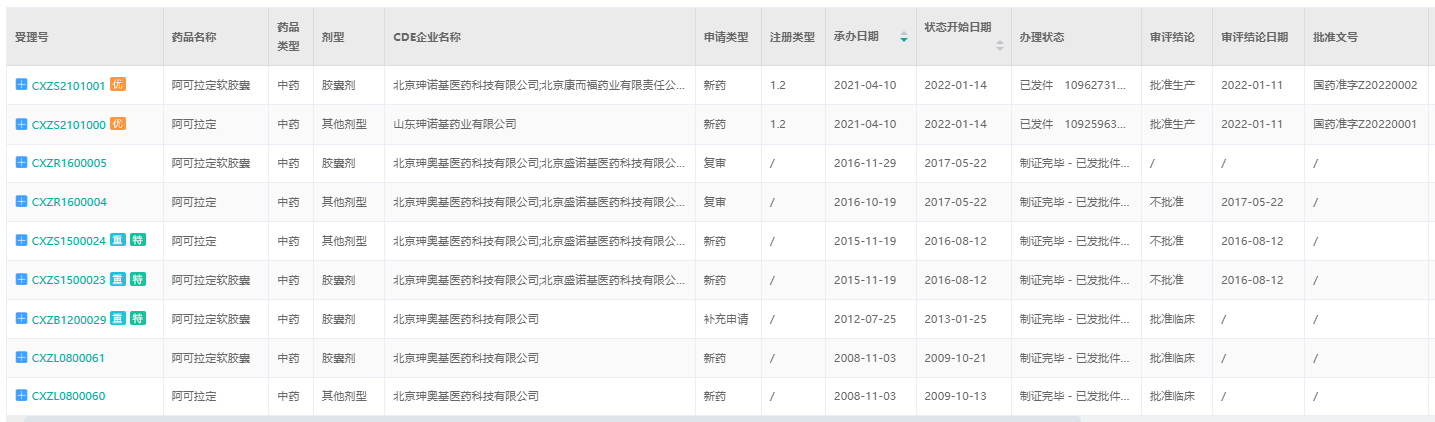
截图来源:药融云中国药品审评数据库
淫羊藿素软胶囊(CXZS2101001)申请上市技术审评报告
一、基本信息
(一)申请人信息
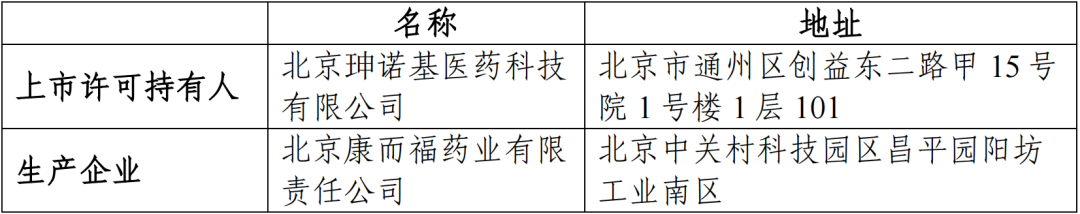
(二)药品的信息
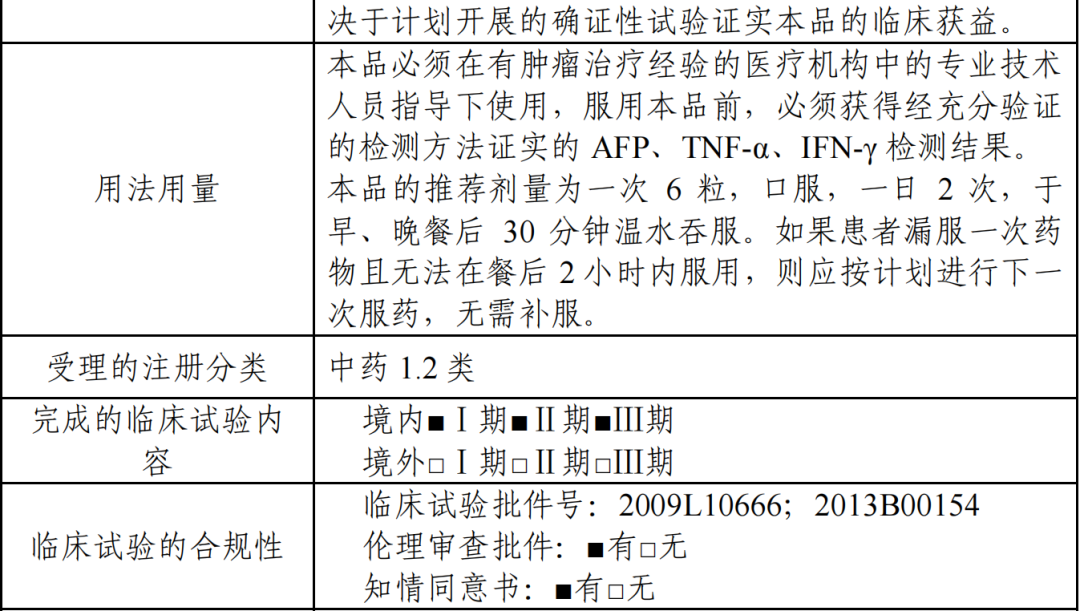
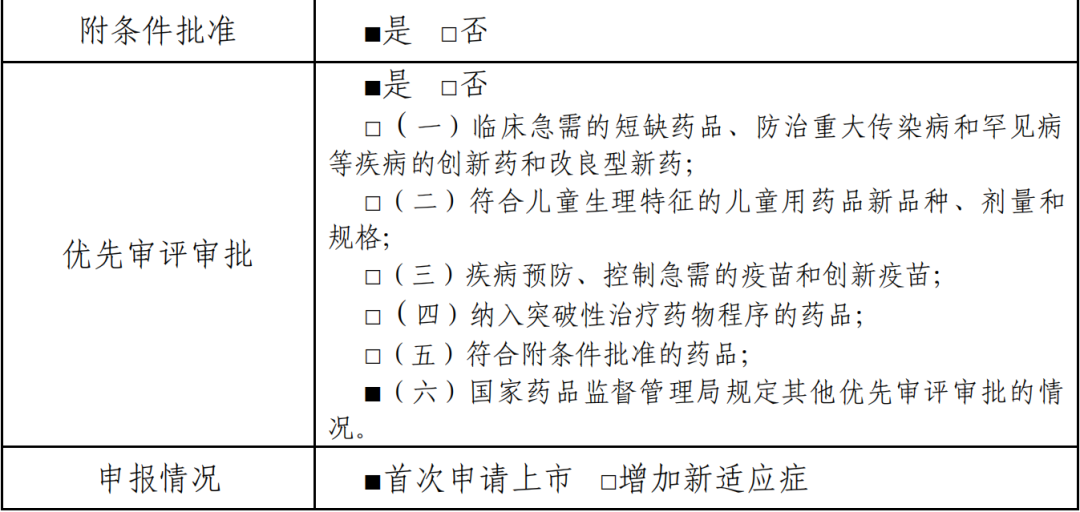
(三)审评经过
受理日期:2021 年 04 月 08 日
召开会议情况


补充资料情况:2021 年 10 月 21 日发出补充资料通知,2021 年 11 月 30日补回资料。
沟通交流情况:2021 年 3 月 12 日、2021 年 11 月 12 日召开了视频沟通交流会。
(四)其他
本品申报临床和申报生产时名称为“阿可拉定软胶囊”。经国家药典委员 会核准,本品通用名称为“淫羊藿素软胶囊”。
二、核查检验及合规评价情况
(一)研制和生产现场检查情况
临床核查:国家药监局核查中心联合广东省和云南省药监局对本品进行 了药品注册临床试验现场核查,被核查单位为南方医科大学南方医院和云南 省中医医院,核查时间为 2021 年 10 月 12 日至 14 日。根据审核意见及核查报告,本品临床试验现场核查未发现真实性问题和重点关注问题。
药学核查:国家药监局核查中心核查组于 2021 年 8 月 17 日至 20 日对本品进行了药品注册现场核查,研制现场和生产现场未发现真实性问题,核查 结论为“通过”。
(二)样品检验情况
中国食品药品检定研究院对三批样品(批号:20201201、20201202、20201203)进行标准复核和样品检验,列入标准的方法可行,样品检验结果 均符合规定。
(三)合规性评价
药审中心基于风险评估启动了本品临床试验数据核查及药学研制/生产现场核查。
本品临床核查未发现真实性问题,药学核查结论为“通过”。合规专业对 现场核查发现的规范性问题进行了合规评价和风险评估,结论为通过。
三、综合审评意见
(一)适应症
本品适用于不适合或患者拒绝接受标准治疗,且既往未接受过全身系统 性治疗的、不可切除的肝细胞癌,患者外周血复合标志物满足以下检测指标 的至少两项:AFP≥400 ng/mL;TNF-α<2.5 pg/mL;IFN-γ≥7.0 pg/mL。
肝细胞癌(hepatocellular carcinoma, HCC)是全球第六大常见癌症,为癌症相关死亡原因的第四位。中国 HCC 病例占全球病例的 55%,为我国致死率第三位的高发肿瘤。目前临床上有相当一部分晚期 HCC 患者因体质较弱、肝功能较差及病情较重等多种原因,不适合采用现有的靶向治疗、系统化疗及 免疫治疗,存在一定的未被满足的临床需求。
(二)药理毒理评价
药理作用:非临床药效学试验结果显示,淫羊藿素对肝癌原位肿瘤模型 小鼠有一定的抑瘤作用,可抑制人肝癌细胞的体外增殖。
毒理研究:
遗传毒性:淫羊藿素 Ames 试验、CHL 细胞染色体畸变试验和小鼠微核试验结果均为阴性。
生殖毒性:大鼠生育力与早期胚胎发育毒性试验、大鼠与兔胚胎-胎仔发 育毒性试验、大鼠围产期生殖毒性试验结果均未见与淫羊藿素相关的生殖毒性。
致癌性:淫羊藿素尚未进行致癌性试验。
其他:Beagle 犬经口给予淫羊藿素 20、60、180 mg/kg,连续给药 9 个月,中、高剂量组可见个别雌性动物乳头及乳腺增大,组织病理学检查可见腺体、 组织增生,且有一定剂量相关性;高剂量组动物 9 个月心电图检查发现 4/8 只动物出现 T 波倒置,ST 段降低。
(三)临床药理学评价
吸收
晚期乳腺癌患者口服淫羊藿素软胶囊后,血浆中药物主要以葡萄糖醛酸 结合代谢产物形式存在,原型药物浓度低;血浆中加β-glucuronidase,把主要 代谢产物水解成原型药物后检测原型药物浓度。
晚期乳腺癌患者餐后单次口服 8 粒淫羊藿素软胶囊,血浆中药物达峰时间(Tmax)为 3.0-6.0 h,峰浓度(Cmax)为 17.9-363.1 ng/mL,餐后单次服用16 粒淫羊藿素软胶囊后 Tmax 为 2.0-4.0 h,Cmax 为 112.2-395.6 ng/mL。
晚期乳腺癌患者餐后口服 2 粒淫羊藿素软胶囊,每日一次连续口服 28 天, 平均稳态峰浓度 Css(max)为 57.5 ng/mL,平均稳态谷浓度 Css(min)为 5.3 ng/mL, 蓄积比(AUC0-t 比值)为 0.9-5.4。
晚期乳腺癌患者餐后单次口服 8-16 粒淫羊藿素软胶囊,与空腹给药相比AUC0-t 提高 2.1-6.9 倍。
分布
晚期乳腺癌患者餐后单次口服 8-16 粒淫羊藿素软胶囊,分布容积 Vz/F 为1935-65152 L。在 0.2-200 μg/mL 浓度范围内,体外淫羊藿素与人血浆蛋白结合率为 82.7-61.3%。
代谢
健康受试者单次口服[14C]淫羊藿素后,血浆中主要检测到 3、7-双葡萄糖醛酸结合和 7-葡萄糖醛酸结合代谢产物。
消除
晚期乳腺癌患者餐后单次服用 8-16 粒淫羊藿素软胶囊后,表观清除率CL/F 为 262-6107 L/h,消除半衰期为 2.0-7.4 h。
健康受试者餐后单次口服[14C]淫羊藿素后,0-96 h 从尿液和粪便中平均总放射性回收率占给药量的 100.62%,其中大部分通过粪便排泄(占给药量的 100.32%),粪便中原型药物占 91.8%,极少量通过尿液排泄(占给药量的0.30%)。
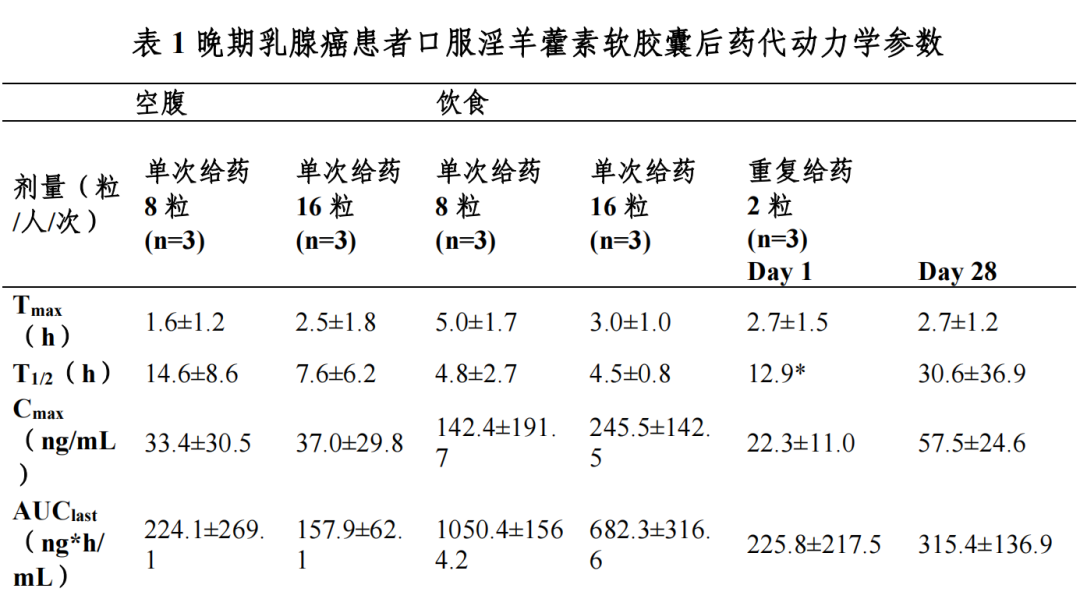
*:统计数据样本量 n=2
在 I 期临床研究中选择了晚期乳腺癌患者进行药代动力学研究,在目标适应症患者中尚未进行药代动力学研究,后续研究正在计划中;后续研究中 继续优化定量检测方法,进行药代动力学研究。
(四)有效性评价
本品无中医药理论及人用经验支持。
1. 关键临床试验设计和结果
2010 年 11 月至 2021 年 11 月开展了Ⅰ期临床试验、ⅠB(ⅡA)期临床试验、ⅡB 期临床试验和Ⅲ期临床试验。ⅡA 和ⅡB 期临床试验为单臂临床试验,Ⅲ期临床试验采用多中心、随机、双盲双模拟、华蟾素片平行对照设计, 试验设计样本量 312 例(富集人群 130 例)。
Ⅲ期临床试验纳入人群为:
年龄≥18 周岁;
符合《原发性肝癌诊疗规范》(2017 年版),临床诊断标准和/或经过病理组织/细胞学检查确诊的晚期或已经发生转移的肝细胞癌(HCC)患者,不 能够采用肝脏手术和/或其他局部治疗(消融或肝动脉介入),或者手术和/或 其他局部治疗后复发进展;
先前未接受过针对晚期或已经发生转移的 HCC 的一线系统治疗;
不适合采用《原发性肝癌诊疗规范》(2017 年版)推荐的晚期肝癌一线标准治疗;
按照实体瘤反应评估标准(RECIST 1.1),至少具有一个可测量靶病灶;
Child-Pugh 肝功能评分 A 级或较好的 B 级(≤7 分);
体力状况 ECOG 评分为 0-1;
预期生存时间≥12 周;
主要器官功能符合以下要求:
①造血功能:血小板≥60×109/L,血红蛋白≥85g/L,白细胞≥3.0×109/L;以上三项可由主要研究者全面衡量患者的状况,适当放宽为:血小板 50-60×10 9/L,血红蛋白 80-85g/L,白细胞 2.5-3.0×10 9/L(含临界值);
②肝脏功能:总胆红素≤1.5 倍正常值上限(ULN),天门冬氨酸氨基转移酶和丙氨酸氨基转移酶≤5×ULN,白蛋白≥28g/L;
③肾脏功能:血清肌酐≤1.5×ULN,或肌酐清除率≥50mL/min;
若HBV-DNA≥104copies/mL(2000IU/mL),必须先行抗病毒保肝治疗,待 HBV-DNA<104copies/mL(2000IU/mL)方可入组;并且继续服用抗病毒药物、监测肝功能和乙肝病毒载量。
富集人群的定义:外周血复合标志物评分≥2 分的患者(即满足以下检测项要求中的两项或三项):①AFP≥400ng/mL(1 分)②TNF-α<2.5 pg/mL(1 分)③IFN-γ≥7.0pg/mL (1 分)。
Ⅲ期临床试验给药方法:试验组服用淫羊藿素软胶囊及华蟾素片模拟剂,华蟾素组服用华蟾素片和淫羊藿素软胶囊模拟剂。淫羊藿素软胶囊/模拟剂用 法为口服,一次 6 粒(每粒装 0.4g,含淫羊藿素 100mg),一日 2 次,连续服用,直至达到终止治疗标准。华蟾素片/模拟剂用法为口服,一次 4 片(0.3g/片),一日 3 次。
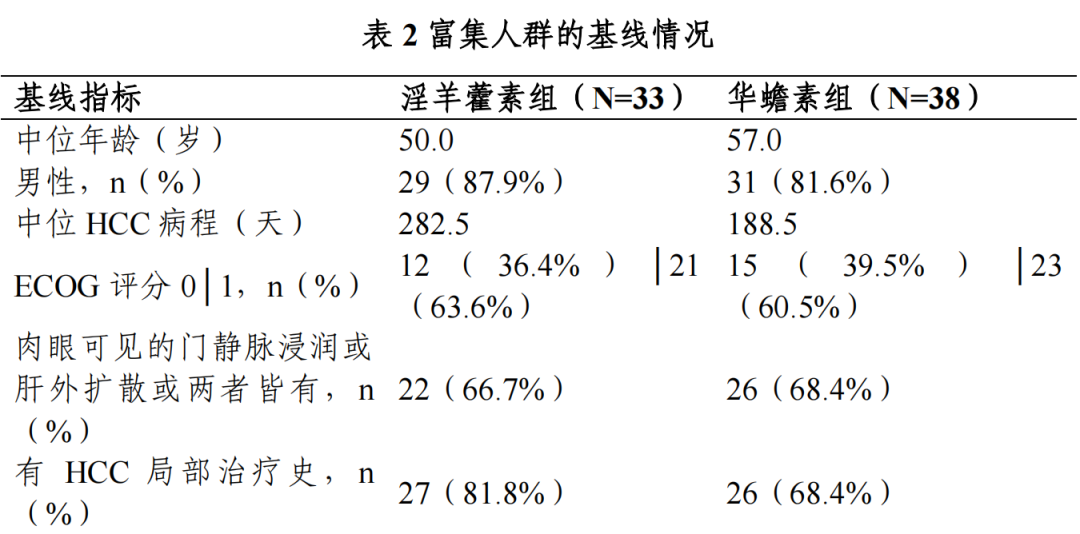
共纳入全人群 282 例,其中淫羊藿素组 141 例,华蟾素组 141 例;富集人群 71 例,其中淫羊藿素组 33 例,华蟾素组 38 例。
Ⅲ期临床试验期中分析富集人群的基线:


Ⅲ期临床试验期中分析有效性结果:
主要疗效指标总生存期(OS)
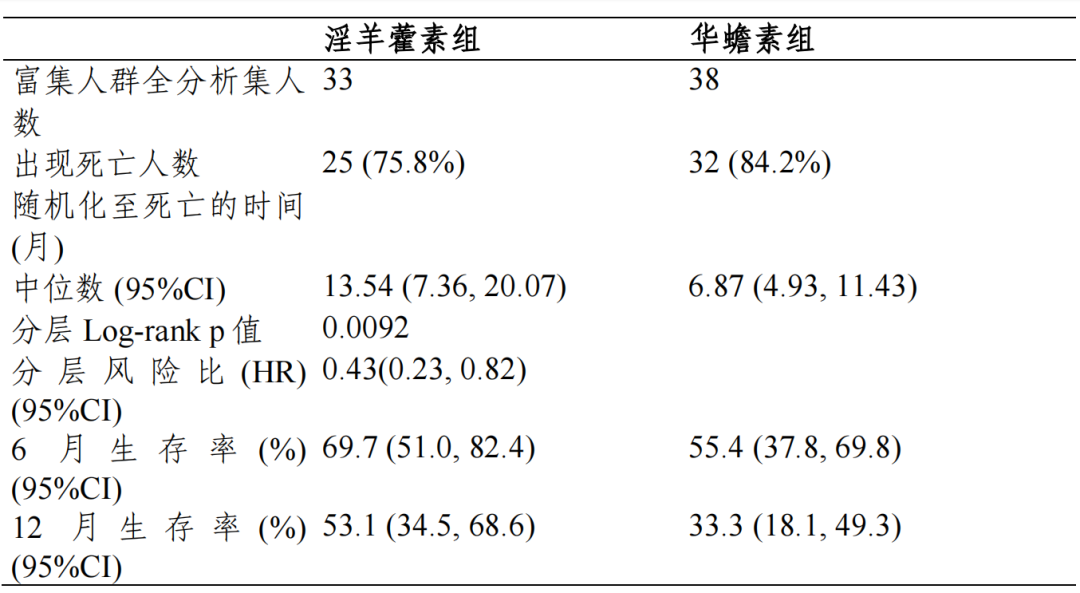
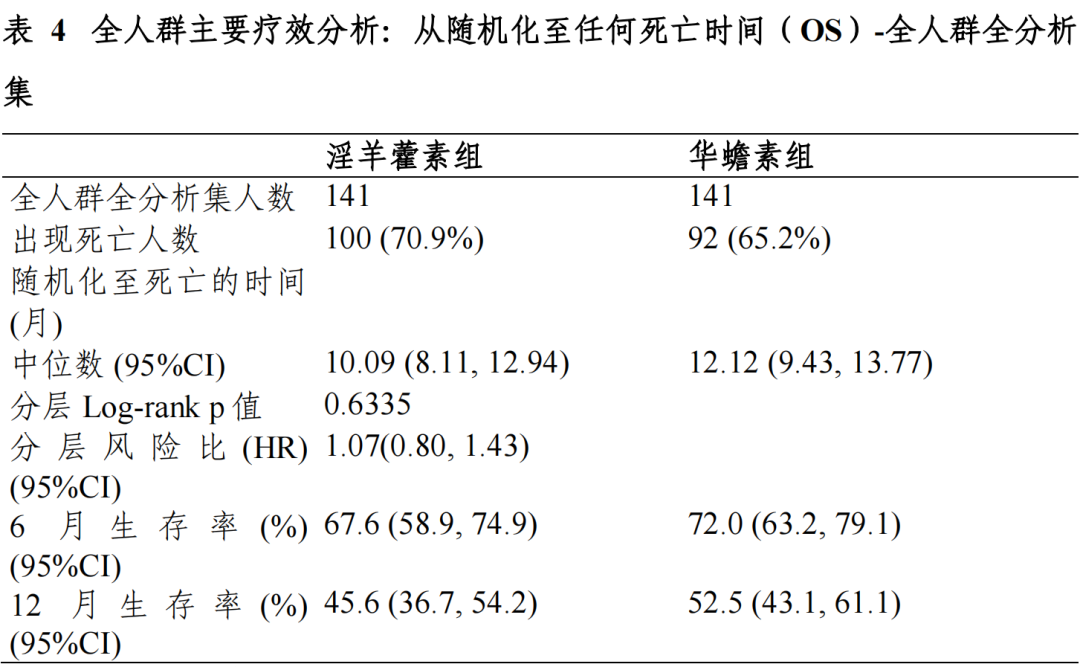
富集人群全分析集:淫羊藿素组和华蟾素组的中位 OS 及 95%CI 分别为13.54(7.36,20.07)个月和 6.87(4.93,11.43)个月,分层风险比(HR)及95%CI 为 0.43(0.23,0.82),p=0.0092,组间差异具有统计学意义。全人群全分析集:两组中位 OS 及 95%CI 分别为 10.09(8.11,12.94)个月和 12.12(9.43,13.77)个月,分层 HR 及 95%CI 为 1.07(0.80,1.43),p=0.6335, 组间差异具有统计学意义。符合方案分析集与全分析集结论一致。

次要疗效指标:
疾病进展时间(TTP)
富集人群全分析集:独立影像评估结果,淫羊藿素组和华蟾素组的中位 疾病进展时间(mTTP)及 95%CI 分别为 1.87(1.84,3.71)个月和 1.84(1.81,1.87)个月,分层 HR 及 95%CI 为 0.63(0.34,1.18),p=0.1031。
全人群全分析集:独立影像评估结果,淫羊藿素组和华蟾素组的 mTTP及 95%CI 分别为 1.87(1.84,3.61)个月和 1.87(1.84,3.65)个月,分层 HR及 95%CI 为 1.06(0.78,1.43),p=0.6955。
无进展生存期(PFS)
富集人群全分析集:独立影像评估结果,淫羊藿素组和华蟾素组的中位无进展生存期(mPFS)及 95%CI 分别为 1.9(1.8,3.7)个月和 1.8(1.8,1.9) 个月,分层 HR 及 95%CI 为 0.72(0.40,1.29),p=0.2038。
全人群全分析集:独立影像评估结果,淫羊藿素组和华蟾素组的 mPFS 及 95%CI 分别为 1.9(1.8,2.0)个月和 1.9(1.9,3.7)个月,分层 HR 及95%CI 为 1.12(0.84,1.47),p=0.4380。
客观缓解率(ORR)
富集人群全分析集:独立影像评估结果,淫羊藿素组和华蟾素组的 ORR及 95%CI 分别为 0.0(0.0%,10.6%)和 2.6%(0.1%,13.8%),p=0.3311。
全人群全分析集:独立影像评估结果,淫羊藿素组和华蟾素组的 ORR 及95%CI 分别为 1.4%(0.2%,5.0%)和 2.8%(0.8%,7.1%),p=0.3834。
疾病控制率(DCR)
富集人群全分析集:独立影像评估结果,淫羊藿素组和华蟾素组的 DCR 及 95%CI 分别为 36.4%( 20.4%, 54.9%)和 23.7%( 11.4%, 40.2%), p=0.3853。
全人群全分析集:独立影像评估结果,淫羊藿素组和华蟾素组的 DCR 及95%CI分别为34.8%(26.9%,43.2%)和36.9%(28.9%,45.4%),p=0.6870。
2. 临床与统计评价
支持本品上市的关键Ⅲ期临床试验“阿可拉定对比华蟾素一线治疗晚期肝 细胞癌受试者的有效性与安全性的多中心、随机、双盲、双模拟、Ⅲ期临床试验”(方案号:SNG1705ICR-1)自 2017 年 9 月 8 日启动,研究原计划入组280 例受试者。在 3.0 版方案生效后,富集人群(复合标志物评分≥2 分)和非富集人群继续按 1:1 随机入组到试验组与对照组。当入组病例数达到 280 例时,若在富集人群中尚未观察到 106 例的死亡事件数,则按 1:1 继续入组富集人群。方案设计基本可行。
对于主要疗效终点 OS,EFAS 富集人群中,淫羊藿素组和华蟾素组的mOS 分别为 13.54(7.36,20.07)个月和 6.87(4.93,11.43)个月,淫羊藿素组的 mOS 相对于华蟾素组显著延长,达到 6.67 个月,分层 HR 及 95%CI 为0.43(0.23,0.82),p=0.0092,组间差异具有统计学意义,亚组分析和根据 研究期间的抗肿瘤治疗分组分析均可支持富集人群相对华蟾素获益的结论, 富集人群有效性结论相对稳健。
(五)安全性评价
1. 安全性数据
本品Ⅰ、Ⅱ、Ⅲ期临床试验,淫羊藿素组共有 269 例受试者进入安全性数据集。
临床试验期间,常见不良反应(≥1%):
胃肠系统疾病:腹泻、恶心、腹胀、呕吐、腹痛、便秘、口干、胃肠出血。
代谢及营养类疾病:食欲减退、低磷酸血症。
肾脏及泌尿系统疾病:蛋白尿、血尿症。
全身性疾病及给药部位各种反应:乏力、发热、体重降低、盗汗、多汗。
心脏器官疾病:室上性期外收缩、室性期外收缩。
血液及淋巴系统疾病:贫血。
皮肤及皮下组织类疾病:瘙痒症、皮疹、湿疹。
呼吸系统、胸及纵隔疾病:鼻衄。
各类神经系统疾病:味觉障碍。
各类检查:天门冬氨酸氨基转移酶升高、丙氨酸氨基转移酶升高、血胆 红素升高、血碱性磷酸酶升高、γ-谷氨酰转移酶升高、血乳酸脱氢酶升高、 血尿酸升高、血小板计数降低、淋巴细胞计数降低、白细胞计数降低、中性 粒细胞计数降低、尿中尿胆原增加、心电图 T 波异常。
≥3 级不良反应:
天门冬氨酸氨基转移酶升高、血胆红素升高、γ-谷氨酰转移酶升高、低 磷酸血症、低钾血症、血小板计数降低、淋巴细胞计数降低、贫血、食欲减 退、尿蛋白检出、尿中尿胆原增加、血压升高、高血压、上呼吸道感染。
≥3 级不良事件:
胃肠系统疾病:腹痛、上消化道出血、上腹痛、腹水、胃肠出血、腹胀、 恶心、肠梗阻、胃底静脉曲张破裂出血、腹内积液。
代谢及营养类疾病:低钠血症、低钾血症、食欲减退、低磷酸血症、高 血糖症、高钙血症、高钾血症。
全身性疾病及给药部位各种反应:死亡、疼痛、多器官功能不全综合征、 乏力、疾病进展、心源性猝死。
血液及淋巴系统疾病:贫血、弥散性血管内凝血。
血管与淋巴管类疾病:出血性休克、高血压。
呼吸系统、胸及纵膈疾病:口咽疼痛、肺栓塞、呼吸困难。
感染及侵染类疾病:上呼吸道感染、胆道感染、慢性乙型肝炎、胃肠炎。
各类神经系统疾病:大脑出血、肝性脑病、昏迷、脊髓压迫、癫痫。
皮肤及皮下组织类疾病:黄色皮肤。
肝胆系统疾病:眼黄疸、胆囊炎、肝肾综合征、肝衰竭、肝细胞性黄疸、 高胆红素血症、肝脓肿。
良性、恶性及性质不明的肿瘤(包括囊状和息肉状):肿瘤出血、癌症 疼痛、肿瘤破裂、脊柱转移、进展性肿瘤。
各类损伤、中毒及手术并发症:道路交通事故、多发性骨折、股骨颈骨折。
眼器官疾病:溃疡性角膜炎。
各种肌肉骨骼及结缔组织疾病:背痛、肌痛。
各类检查:血胆红素升高、天门冬氨酸氨基转移酶升高、γ-谷氨酰转移酶升高、血碱性磷酸酶升高、淋巴细胞计数降低、血小板计数降低、丙氨酸 氨基转移酶升高、体重降低、白细胞计数降低、血压升高、蛋白尿、尿中尿 胆原增加、尿酮体存在、血肌酐升高、血尿酸升高、脂肪酶升高、国际标准 化比率升高、活化部分凝血活酶时间延长、纤维蛋白降解物升高、血甘油三 酯升高、血白蛋白降低、血钙升高。
严重不良事件:代谢及营养类疾病:低钠血症、高钙血症、高钾血症、高血糖症。
各类损伤、中毒及手术并发症:道路交通事故、多发性骨折、股骨颈骨折。
胃肠系统疾病:上消化道出血、肠梗阻、胃肠出血、胃炎、腹内积液。感染及侵染类疾病:胆道感染、感染性肺炎、胃肠炎、脓毒症、肺部感染。
各类神经系统疾病:大脑出血、昏迷、颅内出血、肝性脑病、脊髓压迫、癫痫。
良性、恶性及性质不明的肿瘤(包括囊状和息肉状):肿瘤出血、脊柱 转移、进展性肿瘤。
肝胆系统疾病:肝细胞性黄疸、肝肾综合征、肝衰竭、高胆红素血症、 肝脓肿、肝功能异常。
全身性疾病及给药部位各种反应:死亡、多器官功能不全综合征、发热、 疾病进展、心源性猝死。
眼器官疾病:溃疡性角膜炎。
各种肌肉骨骼及结缔组织疾病:肌痛。
呼吸系统、胸及纵膈疾病:肺栓塞、呼吸困难。血管与淋巴管类疾病:出血性休克。
各类检查:血小板计数降低。
2. 临床与统计评价
主要为1/2 级的胃肠道反应、血常规、肝肾生化指标异常;常见的(发生率≥5%)不良反应主要为天门冬氨酸氨基转移酶升高、血胆红素升高、腹泻、 血小板计数降低、食欲减退、丙氨酸氨基转移酶升高、血碱性磷酸酶升高、蛋白尿、白细胞计数降低、γ-谷氨酰转移酶升高、恶心、乏力;≥3 级不良反应主要为 γ-谷氨酰转移酶升高、血胆红素升高和天门冬氨酸氨基转移酶升高等。
(六)风险分析与控制
本品临床试验期间发现的不良反应和重要不良事件均已在说明书进行风 险提示。对于肝功能异常、肾功能异常、胃肠系统疾病、血小板计数偏低/凝 血功能异常/贫血/低磷酸血症、心肌缺血或心肌梗塞、乳腺增生或子宫内膜增 生、乙肝病毒载量≥104 copies/mL(2000 IU/mL)、妊娠期妇女及哺乳期妇女用药、避孕和儿童用药的注意事项进行了提示。
申请人认为,虽然现有生殖毒性试验结果均未见与本品相关的生殖毒性, 但基于患者和子代安全保护的考虑,对于具有生育能力的妇女和男性,建议采取有效的避孕措施。
制定了风险控制计划,在上市后的确证性临床试验中进一步考察本品的安全性。
(七)获益与风险评估
本品工艺基本稳定可行,基本建立了全过程质量控制体系;现有药效学 结果显示可抑制人肝癌细胞的体外增殖,对拟定适应症有一定的提示作用; 非临床安全性研究基本符合用于恶性肿瘤药物的上市要求。关键性临床试验 纳入的受试者主要为“不适合或不愿意接受标准治疗、且既往未接受过全身系 统性治疗的、不可切除的肝细胞癌”患者,属严重危及生命且尚无有效治疗手 段的疾病。已有Ⅲ期临床试验期中分析结果显示,富集人群当中,本品与对 照药物华蟾素片相比在总生存期上具有一定优势。结合专家审评意见,本品 基本满足《药品附条件批准上市技术指导原则》(试行)的相关要求,综合 评估针对目标人群获益大于风险,具有临床价值。申请人承诺在附条件批准 上市后 4 年内完成相关研究,进一步确证本品的安全性、有效性和人体药代动力学行为。
药审中心与申请人讨论确定了上市后确证性临床试验方案,制定了风险 控制计划。另外,申请人提交了真实世界临床研究等的研究方案。
(八)说明书审核
以Ⅲ期临床试验期中分析的有效性数据和Ⅰ、Ⅱ、Ⅲ期临床试验的安全 性数据为主,结合临床药理学研究的情况,经专家咨询会讨论、中心审评完 成了说明书审核。
关于本品适应症的确定:
本品Ⅲ期临床试验纳入的受试者为:不能够采用肝脏手术和/或其他局部治疗(消融或肝动脉介入),或者手术和/或其他局部治疗后复发进展的肝细胞癌患者,以及不适合采用或者拒绝接受晚期肝细胞癌一线标准治疗的患者。 所有受试者均未接受过针对肝细胞癌的一线系统治疗。
Ⅲ期临床试验期中分析的有效性数据显示,在富集人群(患者外周血复合标志物满足以下检测指标的至少两项:AFP≥400 ng/mL;TNF-α<2.5 pg/mL;IFN-γ≥7.0 pg/mL)当中,本品与对照药物华蟾素片相比在总生存期上具有一定优势,在全人群和非富集人群当中均未展现出有统计学意义的生存获益。结合Ⅲ期临床试验受试人群特征及期中分析有效性结果,经专家咨询会专家审定,确定本品的适应症为:“本品适用于不适合或患者拒绝接受标准治疗, 且既往未接受过全身系统性治疗的、不可切除的肝细胞癌,患者外周血复合标志物满足以下检测指标的至少两项:AFP≥400 ng/mL;TNF-α<2.5 pg/mL;IFN-γ≥7.0 pg/mL。”
四、处理意见
(一)技术结论
经风险获益评估,现有研究和数据支持本品上市用于不适合或患者拒绝 接受标准治疗、且既往未接受过全身系统性治疗的、不可切除的肝细胞癌, 患者外周血复合标志物满足以下检测指标的至少两项:AFP≥400 ng/mL;TNF-α<2.5 pg/mL;IFN-γ≥7.0 pg/mL。
本品基于随机对照Ⅲ期临床试验富集人群的期中分析数据获得附条件批 准上市,本适应症的完全批准将取决于计划开展的确证性试验证实本品的临 床获益。
(二)上市后要求
应于 2025 年 12 月 31 日前完成药物临床试验等相关研究,以补充申请方式申报。在临床试验进程中按相关规定及时沟通进展情况,并定期提交药物 研发期间安全性更新报告。
临床方面:按照 2021 年 12 月 20 日与药审中心沟通后的《淫羊藿素软胶囊对比华蟾素片一线治疗基线时病情复杂、预后较差的晚期肝细胞癌,复合 生物标志物选择人群的前瞻性、随机、阳性平行对照、双盲双模拟的多中心 Ⅲ期临床试验》方案(编号 SNG2111-ICR-1,1.1 版本)开展临床试验,继续考察本品在广泛使用条件下的有效性,做好富集指标检测的质量控制,并进 行系统的安全性观察,必要时补充观察相关指标。
临床药理方面:按照 2021 年 11 月 30 日与药审中心沟通后的《淫羊藿素软胶囊在健康人体内药代动力学研究》方案(编号 SNG2111-ICR-2,1.0 版本) 和《淫羊藿素软胶囊在晚期肝细胞癌患者中的剂量-暴露研究》方案(编号SNG2111-ICR-3,1.0 版本)开展临床试验,进而获得足够的临床药理学数据以支持本品剂量-暴露-效应关系的获得。
药学方面:略。
其他:目前拟定富集生物标志物的临床意义尚不十分明确,请参照《生 物标志物在抗肿瘤药物临床研发中应用的技术指导原则》,继续加强机制研 究。
(三)上市后风险控制
加强上市后风险管理,严格按照所提供的上市后风险管理计划落实风险 防控管理的主体责任,并按相关规定要求及时报告本品上市后临床应用以及 临床试验中出现的非预期严重不良反应(SUSAR)。上市后使用中加强主动 的安全性重点监测,按相关法规要求及时提交本品定期安全性报告。
想要解锁更多药物相关信息吗?查询药融云数据库(https://www.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、申报情况、审批情况、最新进展、市场竞争格局、销售情况、市场规模与前景,可否投入仿制与研发!注册立享15天免费试用和虎年首份医药数据大礼包!

<END>


















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论