



























Abstract
双颗粒系统制剂由两种不同功能的组分组成,可以实现差异化血药浓度与保障产品质量的作用。在其开发过程中应关注不同单元粉体学性质的相似性,对工艺参数进行分别考察,并辅料特性,对不同成分进行归属,从而保障产品的溶出与稳定性。
1 绪论
药品研发是一个收益与风险并存的领域,新分子化合物往往具有极高的收益,但也需要承担较高的失败风险与开发成本,仿制药失败风险与开发成本较低,但收益较低,在4+7政策下,不少品种更是需要面临价格的白刃战。在上述方向之外,改良型新药,即含有已知活性成分的新剂型(包括新的给药系统)、新处方工艺、新给药途径、且具有明显临床优势的药品[1],往往具有较好的风险与收益平衡,成为了许多厂家在第一代新分子实体药物后的改良方向
双组分颗粒系统通常是改良型新药中的一种,指的是由承担不同功能的两种颗粒(以及微丸、微片等)所组成的制剂,该制剂的形式可以是颗粒剂、胶囊剂、片剂等。在市场上,双组分颗粒系统主要有以下三个应用方向:(1)同时实现速释与缓释,快速达到血药起效浓度并维持;(2)隔离可能影响药物释放或与活性成分不相容的功能组分,如作为助悬剂的黄原胶以及作为矫味剂的各种香精;(3)通过将不同释放机理或不同药理作用的组分进行混合,如将需要胃部释放与肠道释放的API封装在同一个制剂中,从而达到联合用药的目的。
双组分颗粒系统制剂的开发相对于常规制剂略微复杂,笔者认为,其开发的核心点分为两个方面,首先,两个单元在混合或者进一步压制后的成品需要具有较好的含量与含量均匀度结果;其次,活性成分与辅料在两个单元之间需要合理分配,降低活性成分释放以及稳定性受影响的风险。在本文中,笔者将根据个人经验从(1)粉体学与工艺(2)溶出与相容性的角度进行简单的探讨。
2 双组分颗粒系统的判别
对于仿制药而言,与参比制剂一致是保证药效的最优解,那怎么判别参比制剂是否使用了双组分颗粒系统呢,笔者认为可以从官网资料以及实物两个方向进行判别。
官方资料(如FDA的Chemical Review)中会对产品的制剂组成进行表述,部分产品在该部分会进行披露并公布两个组分的具体组成(图1),此外,原研专利中的权利要求与实施例也可能会有所披露。

图1 FDA官网公布制剂由迟释微丸与辅料颗粒两部分组成
原研公开相关资料往往可遇不可求,如果官方资料未进行披露,可以对参比制剂实物进行显微观察。多数辅料为白色或类白色,因此,当某些组成(如API、黄原胶、香精等)被有意隔离时,从肉眼角度即可发现两种不同颜色的单元存在。此外,如果显微观察发现存在明显大小与形状不一致的单元,也是判别参比制剂存在双组分颗粒系统的有力依据。
如果参比制剂无法判别,自制制剂是否可以按照双组分颗粒系统进行开发呢?笔者认为其中需要依据产品本身的性质进行评估,一方面,是否有足够的理由(如稳定性等)必须开发双组分颗粒系统,另一方面,开发双组分颗粒系统影响体内的风险是否可以接受。如果产品性质可以得到明显改善,对体内的风险又相对较低,即使参比制剂不是,自制制剂开发双组分颗粒系统又有何妨。
3 粉体学与工艺
含量与产品的有效性息息相关[2],是产品性质的重要方面,而粉体学性质研究无疑是保障制剂含量以及其均匀性的必由之路。
双组分颗粒系统由两个单元组成,提高两个单元之间的相似性,即具有相似的密度与大小,是保障其混合均匀度,从而降低含量与含量均匀度风险的重要措施。笔者在研究中发现,针对同一份空白颗粒,不同的含药颗粒即使在堆密度以及粒度分布上存在差异,亦可获得可接受的含量与含量均匀度结果(表1)。笔者在研究中发现,自制含药颗粒(33~36°)与空白颗粒(约32°)休止角相似,但粒度分布含药颗粒的趋势上含药颗粒要大于空白颗粒,该现象可能是导致RSD整体偏高的原因。因此,笔者认为在项目工期允许的基础上,我们仍应该在制剂开发过程中尽可能减小差异,以降低放大后含量与含量均匀度的风险。
表1 不同处方下的粉体学性质与含量结果

由于双组分颗粒系统涉及两个单元,均需要进行工艺放大,因此,为了降低放大过程的风险并提高成功率,在小试过程中应分别摸索两个单元在工艺上的趋势,并通过工艺的调整,保证放大后两个单元依然一致。以湿法制粒为例,湿法制粒过程中的加水量、加液时间;干燥过程中是否需要冷风干燥;整粒过程的筛网大小与转速,均可能导致两个单元的性质差异,放大过程核心点应为颗粒的相似而不是参数的一致,也就是说,不同的单元可以采用不一样的工艺参数。
4 溶出与相容性
根据笔者的经验,双组分颗粒系统需要考虑的产品性质影响包括两个方面,一方面是增稠剂(如黄原胶)与pH调节剂对溶出行为的影响,另一方面是特定辅料(如糖醇类、香精等)对API降解行为的影响。
作为增稠剂的黄原胶在水中溶胀后会形成复杂的双螺旋状大分子,使得溶液粘度增高,可能会包裹药物分子,进而阻碍药物的释放,因此有可能被添加在空白辅料单元中(图2)。为了保证快速释放,某些API可能对局部微环境的pH有所要求,因此其产品中可能含有磷酸氢钠、柠檬酸、碳酸氢钠等成分,但上述成分并不一定存在于含药单元(图2)
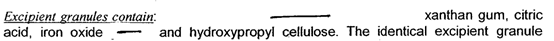
图2 某产品空白颗粒处方组成
许多辅料都可能会与活性成分发生作用[3],其中包括带来美拉德反应的乳糖、含有还原糖杂质的糖醇类辅料、含有氧化杂质的聚维酮等,通过隔离有影响的辅料,双组分颗粒系统在解决上述问题是具有独特的有优势。笔者在处方开发过程中遇到相容性最差的辅料是香精,市售香精中纯香精占比虽然仅占5~10%,但却能显著加速活性化合物的降解,即使全外加亦无法规避。最终,笔者通过将香精加入空白颗粒中解决了稳定性的问题。
此外,值得一提的是,某些原研制剂会对某类功能性辅料(如填充剂)的种类进行专利保护,其可能出发点为同类别的其它辅料无法攻克杂质限度(如糖醇类中的还原糖杂质),在市场上实在无法获得可利用辅料的情况下,开发成双组分颗粒系统,降低API与其接触概率,也是提高产品稳定性的一个有效措施。
5 讨论
在产品开发过程中应对粉体学性质进行全程关注,核心的要求为保证两个单元在性质上的相似性,处方与工艺均可不一致。同时,某些问题通过混合均匀度结果可能并不一定能提前暴露,以颗粒剂为例,在装袋过程中,颗粒下降的高度往往较高,即使是两个休止角都很小的颗粒,也可能会因为设备振动或者下降距离等因素,导致成品的含量均匀度结果较差。而如果是不同的微丸进行压片,两种微丸的可压性也需要进行统一考虑。
针对处方中明确具有相容性问题的组分,通过双组分颗粒系统进行规避,可以大大缩短项目开发的工期。笔者在进行香精筛查时发现,活性成分的降解情况受香精厂家以及种类的影响非常大,甚至不同批次香精在稳定性上表现也略有差异,或许,通过大量的试验我们可以找到合适的香精来源,但所耗费的工期着实无法预期,因此,采用双颗粒系统规避此类问题确实省时省力。
在项目调研中,笔者发现不少原研厂家会选择开发改良型新药,从而获得更长的产品生命周期与市场回报,其改良方向包括开发更有利于口服的胶囊、片剂、颗粒剂,增强药效的缓释剂型等,其中有部分产品就是采用双颗粒组分系统。此外,双颗粒系统的改良方向也可以从患者顺应性角度进行考虑,比如原研口感较差或者单次服用多个药物的便利性等。
6 参考文献
[1] 国家药监局关于发布化学药品注册分类及申报资料要求的通告,2020年第44号.
[2] 口服固体制剂混合均匀度和中控剂量单位均匀度研究技术指导原则(征求意见稿), 2021年09月26日发布.
[3]刘超,宗剑飞, 孟庆廷.化学仿制制剂处方工艺设计与质量控制分析[J]. 中国医药工业杂志,2021, 52(03): 299-311.










 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论