
RAS蛋白是一种由RAS基因编码的低分子量 GDP/GTP 结合蛋白 ,在细胞生长和分化的信号转导中起关键作用。在正常的信号转导过程中,RAS以GTP结合形式执行其功能。然而,由于其低疏水性,RAS本身没有与膜结合的能力。它必须通过酶进行修饰,增强其疏水性,与细胞内膜结合。在发挥其功能后,RAS蛋白被水解成GDP结合形式。RAS突变导致持续的细胞生长信号失控,从而导致细胞过度分化和增殖,最终导致肿瘤发生。RAS(KRAS、NRAS 和 HRAS)是癌症中最常见的突变基因家族,因此,抑制RAS蛋白的活性可以阻止细胞信号转导,是抗癌药物重要的靶点之一。

图1. ICE已构建的RAS家族的生化和SPR实验
A: 生产流程
KRAS[G12D]在HEP E.coli 高表达大肠杆菌中进行表达。

图2. KRAS[G12D]生产流程图
B: 常规QC结果
SDS-PAGE检测KRAS[G12D]的纯度在90%以上,SEC-HPLC检测KRAS[G12D]为单体。
图3. KRAS[G12D] SDS-PAGE(左)和SEC-HPLC(右)结果图
C: 活性验证结果
HTRF法分别进行exchange assay和binding assay对KRAS[G12D]活性验证,KRAS[G12D]具有良好的活性,满足实验需求。
图4. HTRF法活性测定
SPR法对KRAS[G12D]活性验证,KRAS[G12D]满足SPR实验需求。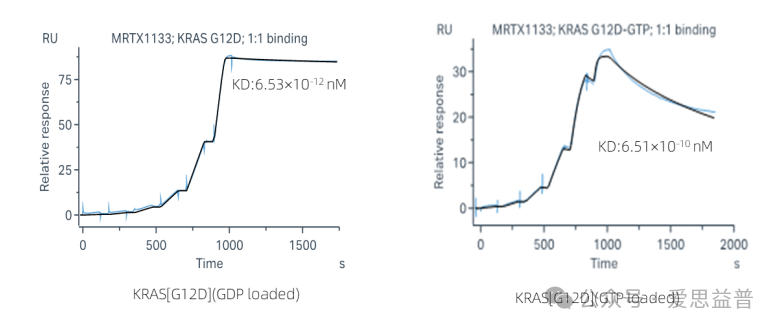
图5. SPR法活性测定

注:RAS系列蛋白产品可提供load GDP、GTP、GMPPNP、GppNHp和生物素化等服务,满足您不同的实验需求。
更多产品信息请访问爱思益普蛋白商城查询:https://protein.ice-biosci.com/;
除了丰富的现货产品,同时可以提供克隆-表达-纯化-质控-活性测定一体化蛋白定制服务。以满足您对蛋白序列、标签或修饰以及不同应用场景的个性化需求;[ 滑动查看更多 ]
Ras蛋白是一种很小的GTP酶,在人类中包含三个RAS原癌基因,如HRAS、KRAS和NRAS,它们是小鼠中HRAS(Harvey 大鼠肉瘤病毒癌基因)、KRAS(Kirsten 大鼠肉瘤病毒癌基因)和NRAS(神经母细胞瘤ras癌基因)的直系同源物。RAS基因参与各种关键细胞信号通路的调节,并调节重要的细胞功能,如增殖、分化、迁移和凋亡等。RAS通过在与活性GTP结合和非活性GDP结合构象状态之间的循环来充当分子开关。这个循环是由GTP酶激活蛋白(GAPs)和核苷酸交换因子(GEFs)调节的。RAS的突变破坏了这一过程并导致组成性激活,从而导致不受控制的细胞生长、增殖和癌症。KRAS是三种最常见的人类RAS蛋白之一,其突变是RAS家族中最主要的突变型,在许多类型的人类癌症中已经检测到KRAS突变。KRAS突变主要发生在第12位和第13位密码子上,以及少量的突变发生在第61位密码子上。KRAS突变普遍存在于胰腺癌(90%)、结肠癌(40%)和肺癌(30%),并且也存在于甲状腺癌、卵巢癌、膀胱癌、全身性红斑狼疮、乳腺癌、肝癌等。NRAS突变存在于黑色素瘤和许多血液系统恶性肿瘤中,HRAS突变主要发生在膀胱癌、甲状腺癌和头颈癌等癌症中。由于KRAS蛋白其光滑的表面和对核苷酸的高亲和力一度被认为是“不可成药”的靶点,直到靶向KRAS G12C共价抑制剂sotorasib和adagrasib的批准上市才打破了KRAS 不可成药的现状。
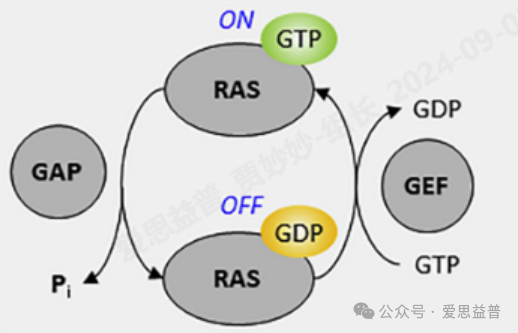
图6. RAS 蛋白的“ON”和“OFF”两种状态
三种RAS基因产生四种主要蛋白质产物:KRAS4A、KRAS4B、NRAS 和 HRAS。这些亚型具有高度同源的序列或结构,并且都具有保守的G结构域 (aa 1-166)和C末端高变区(HVR)(aa 166-188/189)。G结构域包括效应区和变构区,包含核苷酸结合位点以及效应蛋白和调节蛋白的结合位点。效应区包括P环(P-loop,10~17 氨基酸残基)、开关区域 I(Switch I,SWI,30~38 氨基酸残基)、开关区域 II(Switch II, SWII,60~76 氨基酸残基),其中 SWI 区和 SWII 区在RAS 亚型中具有保守性,是介导蛋白-蛋白相互作用的重要区域。变构区由 87~172 氨基酸残基组成,在各种RAS亚型中具有86%的相似性,与蛋白构象分布状态和膜相互作用有关。C端的高变区域(167~188 氨基酸残基)包含CAAX(C,半胱氨酸;A,脂肪族;X,任何氨基酸)序列,依赖多聚赖氨酸区域的静电相互作用实现膜定位,还与KRAS蛋白二聚体的形成和信号转导有关。KRAS蛋白与三磷酸鸟苷(guanidine triphosphate,GTP)/二磷酸鸟苷(guanosine 5'- diphosphate,GDP)结合时会导致SWI和SWII发生构象变换,当蛋白处于和 GTP结合的活化状态时,T35和G50与γ-磷酸形成氢键,此时SWI和SWII处于活性构象,若GTP被水解生成GDP则氢键断开,SWI和SWII转变为非活性构象。活化的KRAS蛋白则形成二聚体或者高价多聚体介导下游信号通路,KRAS 突变蛋白会导致蛋白二聚体构象发生变化,错误的二聚体构象是驱动致癌信号传导的重要诱因。

图7.RAS蛋白的结构示意图
KRAS蛋白由于其结构功能特征一度被认为是“不可成药”靶点。一方面,KRAS蛋白与GTP/GDP具有较高亲和力(p摩尔级),且GTP和GDP在血液中具有较高的浓度;另一方面,KRAS蛋白具有结构复杂,缺乏适宜药物结合口袋的特征。近年来,关于KRAS抑制剂的研究获得突破。2021年5月,美国FDA宣布加速批准选择性KRAS G12C抑制剂sotorasib(AMG510)上市,这是全球首个获得批准的靶向KRAS突变的肿瘤治疗药物。2022年12月,adagrasib(MRTX849)获批上市。这两个药物的批准上市打破了KRAS不可成药的现状。此外,KRAS G12D 的选择性非共价抑制剂MRTX1133也提供了一种新型靶向治疗。在这个靶向KRAS突变的新时代,下一个挑战将是了解和克服耐药机制。

图8. KRAS 突变抑制剂靶向的KRAS表面结构
AMG510(sotorasib)是首个进入临床试验的特异性靶向KRAS G12C的小分子抑制剂,其特异性且不可逆地与诱导型S-IIP 中的Cys12结合,并将KRAS G12C蛋白锁定在非活性状态,.AMG510还与KRAS上His95的替代取向形成的新型表面凹槽相结合,与ARS-1620相比,效率提高了10 倍。令人兴奋的是,AMG510于2021年5月28日获得美国食品药品监督管理局(FDA)批准,作为肿瘤携带 KRAS G12C突变且既往接受过至少一种全身治疗的NSCLC成年患者的首个治疗方法。这是一个里程碑,因为它是第一个直接靶向突变 KRAS 的药物。
Mirati Therapeutics开发的MRTX849(adagrasib)已被确定为一种高选择性的KRAS(G12C)共价抑制剂,目前处于I/II期临床研究中。它是一种口服、小分子选择性的 KRAS (G12C) 突变抑制剂。它不仅可以在体内几乎完全抑制 KRAS 突变,而且具有良好的类药特性。MRTX849选择性靶向 GDP 中 KRAS 的突变半胱氨酸 12,该半胱氨酸存在于 KRAS 的诱导开关 II 口袋 (G12C) 中,从而将其锁定为失活的 GDP 结合状态并抑制 RAS/MAP 激酶通路。在 KRAS (G12C) 阳性细胞系和来自多种肿瘤类型的患者来源的异种移植模型中,65% 的模型显示显着的肿瘤消退。
ARS-1620是基于ARS-853进行结构改造和优化获得的共价抑制剂,是第 一个被证明在体内直接靶向KRAS G12C发挥抗肿瘤增殖活性的小分子抑制剂,弥补了ARS-853药代动力学性质的不足和体内实验数据的缺乏。该化合物特异性作用于KRAS G12C的S-II 口袋,与野生型 KRAS蛋白不发生相互作用。
MRTX1133是Mirati Therapeutics研发的一款选择性非共价 KRAS G12D 抑制剂。临床前研究结果显示:MRTX1133 能选择性与 KRAS G12D 突变体结合,在多种携带 KRAS G12D 突变的肿瘤细胞系中均显示出特异性抑制KRAS依赖的相关信号通路,并且MRTX1133和EGFR单抗西妥昔单抗联合用药相比于单药能够抑制肿瘤细胞pERK和pS6的表达,显著提高抗肿瘤效应。有研究显示,MRTX1133 能够显著抑制KRAS G12D突变的胰腺癌肿瘤生长,可能为胰腺癌患者治疗带来曙光。MRTX1133 目前已经获得了 FDA 批准,获得新药临床试验申请批准,在2023年已经开始进入I/II 期临床阶段。
RMC-6236是一种强效的口服三复合物RAS(ON)小分子抑制剂,旨在治疗由多种RAS突变驱动的癌症。RMC-6236与丰富的细胞内伴侣蛋白亲环蛋白A(CypA)非共价结合,产生与RAS(ON)结合形成高亲和力、RAS选择性三复合物的二元复合物,该复合物在空间上抑制RAS与下游cRAF的结合。暴露于 RMC-6236抑制ERK磷酸化和细胞生长,并在体外诱导多种人RAS成瘾癌细胞系细胞凋亡。RMC-6236在体内促进抗肿瘤免疫,并与抗 PD1抗体相加,在 KRAS 突变CRC模型中驱动持久的完全反应和免疫记忆。此外,RMC-6236 治疗在检查点阻断难治性KRAS突变模型中逆转了致癌RAS驱动的免疫逃避机制,显着改变了肿瘤微环境,有利于抗肿瘤免疫。
约30%的人类肿瘤疾病是由RAS基因突变引起的,在所有的病例中,约85%是由KRAS突变引起的,因此靶向KRAS突变蛋白是治疗癌症的一种重要手段。虽然关于直接抑制KRAS突变蛋白的小分子抑制剂已经取得了一些进展,但目前KRAS G12D和KRAS G12V等常见突变体的小分子抑制剂研究仍然面临着重大挑战,且已获批药物随之而来的耐药性和脱靶毒性等问题也为小分子 KRAS 抑制剂的研究带来了新的困难。然而,随着对KRAS蛋白不断的探索和研究,相信在未来能够获得更好疗效的KRAS抑制剂与最优药物组合应用于临床,造福于广大患者。[1].Reck M, Carbone DP, Garassino M, Barlesi F. Targeting KRAS in non-small-cell lung cancer: recent progress and new approaches. Ann Oncol. 2021 Sep;32(9):1101-1110. doi: 10.1016/j.annonc.2021.06.001. Epub 2021 Jun 2. PMID: 34089836.[2].Takács T, Kudlik G, Kurilla A, Szeder B, Buday L, Vas V. The effects of mutant Ras proteins on the cell signalome. Cancer Metastasis Rev. 2020 Dec;39(4):1051-1065. doi: 10.1007/s10555-020-09912-8. PMID: 32648136; PMCID: PMC7680337.[3].李学燕,陈娜,江程. 靶向KRAS蛋白抑制剂的研究进展[J]. 中国药科大学学报,2024,55(2):257-269. DOI:10.11665/j.issn.1000-5048.2024010801.[4].Zhu C, Guan X, Zhang X, Luan X, Song Z, Cheng X, Zhang W, Qin JJ. Targeting KRAS mutant cancers: from druggable therapy to drug resistance. Mol Cancer. 2022 Aug 4;21(1):159. doi: 10.1186/s12943-022-01629-2. PMID: 35922812; PMCID: PMC9351107. 北京爱思益普生物科技股份有限公司 2010年创建,致力于打造靶点驱动的药物发现生物学平台。作为创新型CRO+的探索者,爱思益普专注于以“新靶点、新方法、新技术”解决创新药从靶点发现到候选化合物确认阶段的生物学和成药性的挑战;同时,爱思益普融合临床医学和生物学的专业团队,基于对疾病生物学及药物研发逻辑的深入理解,建立药物发现“一体化”的生物学平台为客户提供综合解决方案。爱思益普关注新药研发企业对质量、效率和成本的需求,用专业的生物学技术和高效的沟通帮助客户提高新药研发的效率及成功率。
北京爱思益普生物科技股份有限公司 2010年创建,致力于打造靶点驱动的药物发现生物学平台。作为创新型CRO+的探索者,爱思益普专注于以“新靶点、新方法、新技术”解决创新药从靶点发现到候选化合物确认阶段的生物学和成药性的挑战;同时,爱思益普融合临床医学和生物学的专业团队,基于对疾病生物学及药物研发逻辑的深入理解,建立药物发现“一体化”的生物学平台为客户提供综合解决方案。爱思益普关注新药研发企业对质量、效率和成本的需求,用专业的生物学技术和高效的沟通帮助客户提高新药研发的效率及成功率。爱思益普建立的技术平台包括:
1、基于靶点的药物筛选平台:建立了超过1500种药物靶点,涵盖大多数离子通道,GPCR,激酶和非激酶靶点以及近万个实验方法,建立了从生物物理学、生物化学、细胞生物学、电生理学平台以及各种多平台和高通量筛选技术。
2、体外和体内药效筛选评价平台:包括肿瘤、免疫、心血管、中枢神经系统、代谢基于细胞、组织或动物模型的药效学评价。
3、早期成药性筛选评价平台:包括药物发现阶段ADME和PK研究,以及药物脱靶效应筛选(hERG,safety panel,激酶谱等)
4、新分子平台:专注于ADC、多肽、蛋白降解、小核酸、细胞治疗等多种新分子的药物发现研究。





































 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论