
细胞周期蛋白依赖激酶(Cyclin-dependent kinases, CDKs)是蛋白质参与关键细胞过程的激酶,如细胞周期或转录,其发挥功能活性需要与特定的细胞周期蛋白亚基结合。迄今为止,已知有20个CDKs家族成员调节细胞周期、转录和剪接。CDKs通过与周期蛋白结合,确保细胞在正确的时间进行DNA复制和细胞分裂。CDK功能失调与肿瘤发生发展密切相关,CDK是抗肿瘤药物研发中的重要靶点。
爱思益普已表达纯化超30种CDK系列活性蛋白可供选择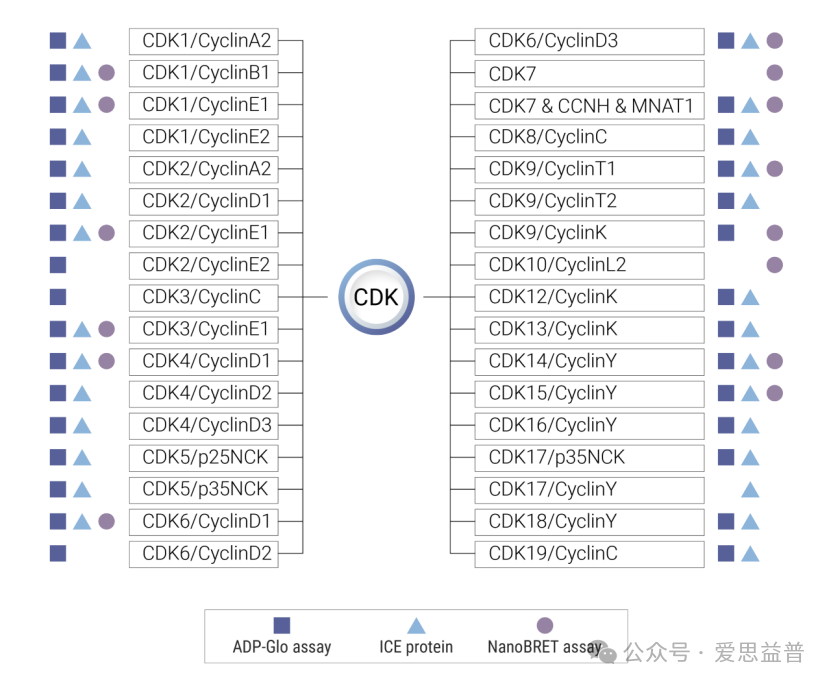
图1. ICE已构建的CDK家族的体外酶活性和细胞测定实验
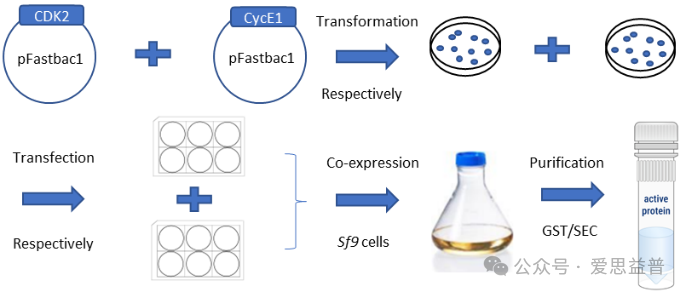
A: 生产流程
CDK2和CycE1在HEP sf9 高表达昆虫细胞中共表达。
B: 常规QC结果
SDS-PAGE检测CDK2/CycE1的纯度在90%以上。图3. CDK2/CycE1 SDS-PAGE图
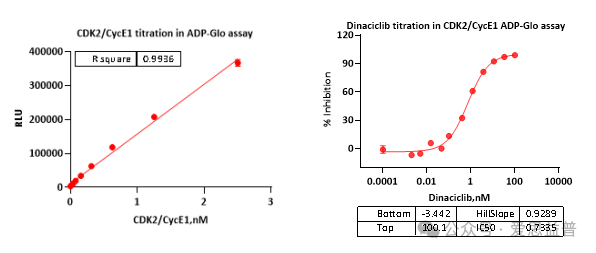
C: 活性验证结果
ADP-Glo法对CDK2/CycE1分别进行酶滴和阳性药的验证,CDK2/CycE1具有良好的活性,满足实验需求。更多产品信息请访问爱思益普蛋白商城查询:https://protein.ice-biosci.com/除了丰富的现货产品,同时可以提供克隆-表达-纯化-质控-活性测定一体化蛋白定制服务。以满足您对蛋白序列、标签或修饰以及不同应用场景的个性化需求。[ 滑动查看更多 ]
细胞周期是指细胞分裂进程的周期性过程,在细胞发育过程中起着至关重要的作用。许多疾病的发生是由于细胞周期调控失衡引起的,如人恶性肿瘤的分子特征是细胞周期调节的失控,引起细胞分化的缺乏和细胞生长的失常,导致细胞无限增殖。而细胞周期蛋白依赖性激酶、周期蛋白是整个细胞周期调控机制中的核心分子。其中,CDK是主要的细胞周期调节因子,可与周期蛋白结合形成周期蛋白- CDK复合物,从而介导底物磷酸化,参与细胞周期的调控。
促有丝分裂信号在细胞周期早期促进细胞周期蛋白D的合成,细胞周期蛋白D结合并激活CDK4/6,随后激活CDK2/cyclin E。CDK4/CDK6使Rb蛋白磷酸化,CDK2进一步使Rb过度磷酸化。Rb的过度磷酸化阻止E2F结合,使细胞进入S期,在那里发生DNA合成。在S期接近结束时,CDK2/细胞周期蛋白A磷酸化E2F1,阻止其与DNA结合并终止S期。CDK1与细胞周期蛋白B偶联完成有丝分裂过程。CDK3与细胞周期蛋白C结合使Rb磷酸化,导致从G0期退出。除了这些直接驱动细胞周期进程的CDK外,还发现了CDK7、CDK8、CDK9、CDK10、CDK11、CDK12、CDK13和CDK19等其他CDK,并证实参与转录调控。作为CDK激活激酶(CAK),CDK7磷酸化CDK1和CDK2等CDKs,也参与细胞周期的调节。
CDKs和Cyclins的过度表达、CDK抑制因子的缺失或功能障碍会导致CDK活性的调控异常,进而引起细胞周期紊乱,导致恶性肿瘤的发生。抑制其催化活性可以对恶性肿瘤等增生性疾病的治疗起到积极作用,因此细胞周期蛋白依赖性激酶(CDKs)抑制剂的研究有着重要意义,是非常有前景的癌症治疗领域。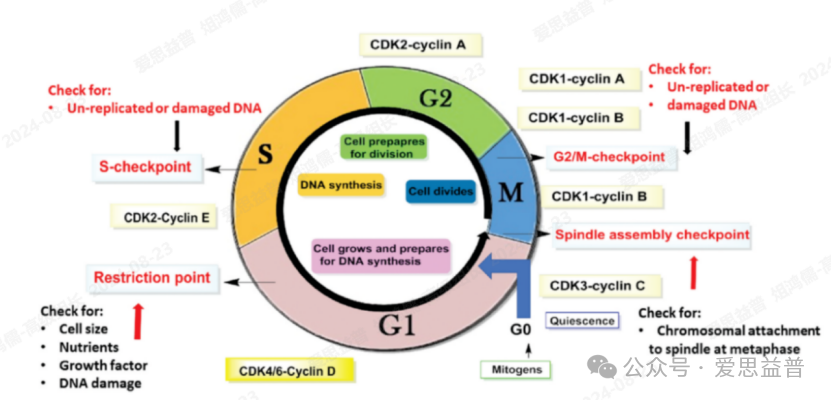
图5. CDKs调控细胞周期
细胞周期蛋白依赖性激酶(cyclin dependent kinase, CDK)是丝氨酸/苏氨酸蛋白激酶家族成员,通过介导不同底物磷酸化,参与细胞周期调控和转录调节。大多数CDK具有CDK激酶结构域、周期蛋白结合位点、磷酸化修饰位点和T-loop (称为激活环)基序等。当CDK与其特定的周期蛋白非共价结合时CDK的T-loop被置换,从而暴露底物ATP结合位点并重新排列活性位点的关键残基,激活CDK活性。细胞周期蛋白依赖性激酶按照功能可以分为两种:(1)与细胞周期相关的CDKs(CDK1-7 和 14-18),可直接调节细胞周期各阶段的进展;(2)与转录相关的CDKs(CDK7-13、19 和 20)。肿瘤细胞增殖通常与遗传和表观遗传机制相关,通常影响细胞周期调节蛋白的表达或导致检查点控制不合格,导致对细胞损伤的异常反应从而促进肿瘤进展。
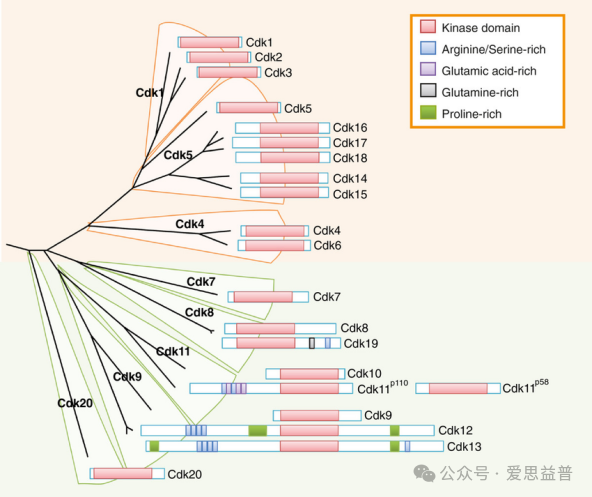
图6.哺乳动物CDK家族
在CDK抑制剂的开发过程中,存在多种困难和误解,包括对CDKs生物学功能的不充分理解,对泛CDK和多CDK抑制剂的关注不足,已开发的选择性CDK抑制剂的非理想亚型选择性,早期发现的CDK抑制剂的代谢稳定性被忽视,缺乏有效的抗药性解决方案,以及缺乏CDK治疗的有效联合疗法和生物标志物。
图7.截止2021年12月在不同临床阶段的CDKs抑制剂
抑制CDKs已成为治疗各种疾病,特别是癌症的有效治疗策略。尽管过去近三十年中在CDK抑制剂的发现上做出了巨大努力,并且许多抑制剂已进入临床试验,但目前仅有五种CDK抑制剂获得批准:哌柏西利(Palbociclib),阿贝西利(Abemaciclib),瑞博西尼(Ribociclib),曲拉西利(Trilaciclib)和达尔西利(Dalpiciclib)。Palbociclib 是一种第一代的 CDK4/6 选择性抑制剂。在2015年,被美国食品药品监督管理局(FDA)批准用于治疗雌激素受体阳性(ER+)的乳腺癌。Abemaciclib,Ribociclib与Palbociclib类似也属于第一代的 CDK4/6 选择性抑制剂,主要用于治疗激素受体阳性(HR+)、人表皮生长因子受体2阴性(HER2-)的晚期或转移性乳腺癌。它们都属于 ATP 竞争性抑制剂,也称为 Type I CDK 抑制剂。Type I CDK 抑制剂通过与 CDKs 的 ATP 结合位点竞争结合,从而阻止 CDKs 与 ATP 的结合,进而抑制 CDKs 的活性。Palbociclib 通过抑制 CDK4/6,导致细胞周期停滞在 G1 阶段,抑制了肿瘤细胞的增殖。Palbociclib 对 CDK4 和 CDK6 有较高的选择性,这有助于减少对其他 CDKs 或激酶的非特异性抑制,从而可能降低某些副作用。palbociclib联合PARP抑制剂olaparib治疗myc驱动的卵巢癌疗效良好。Abemaciclib作为单药治疗或与其他抗癌药物联合使用,已被证明可以提高患者的无进展生存期和总生存期。Trilaciclib 是一种 CDK4/6 抑制剂,但它与传统的 CDK4/6 抑制剂(如 Palbociclib、Ribociclib 和 Abemaciclib)有所不同。Trilaciclib 主要被开发用于减少化疗引起的骨髓抑制,并且它的作用机制更侧重于保护骨髓和免疫系统,而不是直接针对肿瘤细胞的增殖。Trilaciclib属于非ATP竞争性抑制剂,也称为 Type II 抑制剂。与传统的 ATP 竞争性 CDK 抑制剂(Type I)不同,Trilaciclib 通过结合 CDK4/6 的非活性构象来发挥作用,这种结合方式不直接与 ATP 结合位点竞争。2021年2月,Trilaciclib 获得了美国 FDA 的批准,用于减少在接受含铂/依托泊苷方案或拓扑替康方案的广泛期小细胞肺癌(ES-SCLC)成年患者化疗引起的骨髓抑制。另外,Trilaciclib 在中国的 III 期临床试验也在进行中,该试验旨在评价 Trilaciclib 在接受卡铂联合依托泊苷或拓扑替康治疗的广泛期小细胞肺癌患者中的安全性、有效性和药代动力学特征。Dalpiciclib是首个中国国产原研CDK4/6抑制剂,本次获批适应症为联合氟维司群用于激素受体(HR)阳性,人表皮生长因子受体2(HER2)阴性的经内分泌治疗后进展的复发或转移性乳腺癌的治疗。除了上述已获批的CDK4/6抑制剂,还有新型的CDK2选择性降解剂正在研究中。例如,清华大学饶燏团队与浙江大学应美丹团队合作发表了关于CDK2选择性降解剂的设计及其在AML分化治疗中的应用研究,这为AML治疗提供了一种潜在的分化治疗方案。这项研究中的CDK2选择性降解剂,通过靶向CDK2并利用细胞的泛素-蛋白酶体系统诱导其降解,从而达到治疗AML的效果。研究者通过这种新型降解剂,成功诱导了AML细胞的分化,显示出了良好的治疗潜力和较低的毒性,为AML的分化治疗提供了新的希望。新型CDK2抑制剂INCB123667在临床前生化分析中显示出对CDK2的选择性,而对其他CDKs无活性,这可能为CDK4/6抑制剂耐药的癌症患者带来新的治疗选择。此外,CDK2抑制剂BLU-222虽然因眼部不良事件暂停了部分试验,但很快取消了限制,并继续在多种癌症中进行研究。同时,CDK9抑制剂也在研究之中,它们通过抑制CDK9介导的转录延伸阶段,可能对某些类型的癌症具有治疗潜力。此外,CDK12抑制剂的研究也在进展中,CDK12参与基因转录的多个环节,其功能失调与肿瘤发生发展相关,CDK12抑制剂的研究可能为癌症治疗提供新的策略。总体来看,CDK抑制剂的研究和开发正朝着更精准、更有效的方向发展,未来有望为癌症患者提供更多、更好的治疗选择。
1.PAN Jianfeng, SHANG Fangzheng, MA Rong, RONG Youjun, ZHANG Yanjun. Advances of the regulatory mechanism of cyclin, cyclin- dependent kinases and related kinase inhibitors in cell cycle progression[J]. Chinese Journal of Biotechnology, 2023, 39(4): 1525-1547.2.Łukasik P, Załuski M, Gutowska I. Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development-Review. Int J Mol Sci. 2021 Mar 13;22(6):2935.3.Nardone V, Barbarino M, Angrisani A, Correale P, Pastina P, Cappabianca S, Reginelli A, Mutti L, Miracco C, Giannicola R, Giordano A, Pirtoli L. CDK4, CDK6/cyclin-D1 Complex Inhibition and Radiotherapy for Cancer Control: A Role for Autophagy. Int J Mol Sci. 2021 Aug 4;22(16):8391.4.Alshaye NA. Synthesis and invitro anticancer activity of some 2-oxindoline derivatives as potential CDK2 inhibitors. J Biomol Struct Dyn. 2023;41(24):15009-15022.5.Xie Z, Hou S, Yang X, Duan Y, Han J, Wang Q, Liao C. Lessons Learned from Past Cyclin-Dependent Kinase Drug Discovery Efforts. J Med Chem. 2022 May 12;65(9):6356-6389. 北京爱思益普生物科技股份有限公司 2010年创建,致力于打造靶点驱动的药物发现生物学平台。作为创新型CRO+的探索者,爱思益普专注于以“新靶点、新方法、新技术”解决创新药从靶点发现到候选化合物确认阶段的生物学和成药性的挑战;同时,爱思益普融合临床医学和生物学的专业团队,基于对疾病生物学及药物研发逻辑的深入理解,建立药物发现“一体化”的生物学平台为客户提供综合解决方案。爱思益普关注新药研发企业对质量、效率和成本的需求,用专业的生物学技术和高效的沟通帮助客户提高新药研发的效率及成功率。
北京爱思益普生物科技股份有限公司 2010年创建,致力于打造靶点驱动的药物发现生物学平台。作为创新型CRO+的探索者,爱思益普专注于以“新靶点、新方法、新技术”解决创新药从靶点发现到候选化合物确认阶段的生物学和成药性的挑战;同时,爱思益普融合临床医学和生物学的专业团队,基于对疾病生物学及药物研发逻辑的深入理解,建立药物发现“一体化”的生物学平台为客户提供综合解决方案。爱思益普关注新药研发企业对质量、效率和成本的需求,用专业的生物学技术和高效的沟通帮助客户提高新药研发的效率及成功率。爱思益普建立的技术平台包括:
1、基于靶点的药物筛选平台:建立了超过1500种药物靶点,涵盖大多数离子通道,GPCR,激酶和非激酶靶点以及近万个实验方法,建立了从生物物理学、生物化学、细胞生物学、电生理学平台以及各种多平台和高通量筛选技术。
2、体外和体内药效筛选评价平台:包括肿瘤、免疫、心血管、中枢神经系统、代谢基于细胞、组织或动物模型的药效学评价。
3、早期成药性筛选评价平台:包括药物发现阶段ADME和PK研究,以及药物脱靶效应筛选(hERG,safety panel,激酶谱等)
4、新分子平台:专注于ADC、多肽、蛋白降解、小核酸、细胞治疗等多种新分子的药物发现研究。








































 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论