



























在8月,很多跨国药企的收购或合作到了收获期,例如吉利德最近通过43亿美元重金收购CymaBay Therapeutics公司而获得的一款肝病药物可能会获得批准,Syndax和Incyte合作开发的抗CSF-1R单抗可能获得批准。除此之外,首个用于实体瘤适应症的新型TCR-T细胞疗法、首款迷幻药辅助疗法、全球第三款BCMA/CD3双抗也可能上市,这都将给市场带来里程碑意义。
以下是2024年8月值得关注的五项FDA决定:
一、Afami-cel
研发公司:Adaptimmune Therapeutics
PDUFA日期:2024年8月4日
1月31日,Adaptimmune宣布其TCR-T疗法Afami-cel的上市申请获FDA受理并获得优先审评资格,用于治疗晚期滑膜肉瘤,PDUFA日期为2024年8月4日。
艾基奥仑赛(afamitresgene autoleucel,afami-cel)是一种靶向MAGE-A4的TCR-T细胞疗法,如果获得批准,艾基奥仑赛将是首个用于实体瘤适应症的市售工程化T细胞疗法。

此次BLA的递交是基于一项2期SPEARHEAD-1研究(NCT04044768)中队列1的积极数据。该研究评估了单次静脉输注afami cel对HLA-a*02符合条件和MAGE-A4阳性的晚期滑膜肉瘤或粘液样/圆细胞脂肪肉瘤患者在接受淋巴消耗性化疗后的疗效和安全性。入组的受试者既往接受过中位数为3线或3线以上的系统治疗。
该研究达到了主要疗效终点,患者的总缓解率(ORR)约为39%,中位缓解持续时间约为12个月。

Afami-cel临床数据(图源:Adaptimmune官网)
以往接受过两种或两种以上疗法的滑膜肉瘤患者的中位总生存期(OS)小于12个月,而接受afami-cel治疗患者的中位OS约为17个月,优于历史对照数据。接受afami-cel治疗有缓解的晚期滑膜肉瘤患者中,70%的人在治疗两年后仍然存活。

Afami-cel临床数据(图源:Adaptimmune官网)
其他分析表明,较高的afami-cel细胞持久性与较长的OS有关。安全性方面,毒性反应包括细胞因子释放综合征和可逆的血液学毒性,这与之前的研究结果一致,表明afami-cel的安全性是可以接受的。
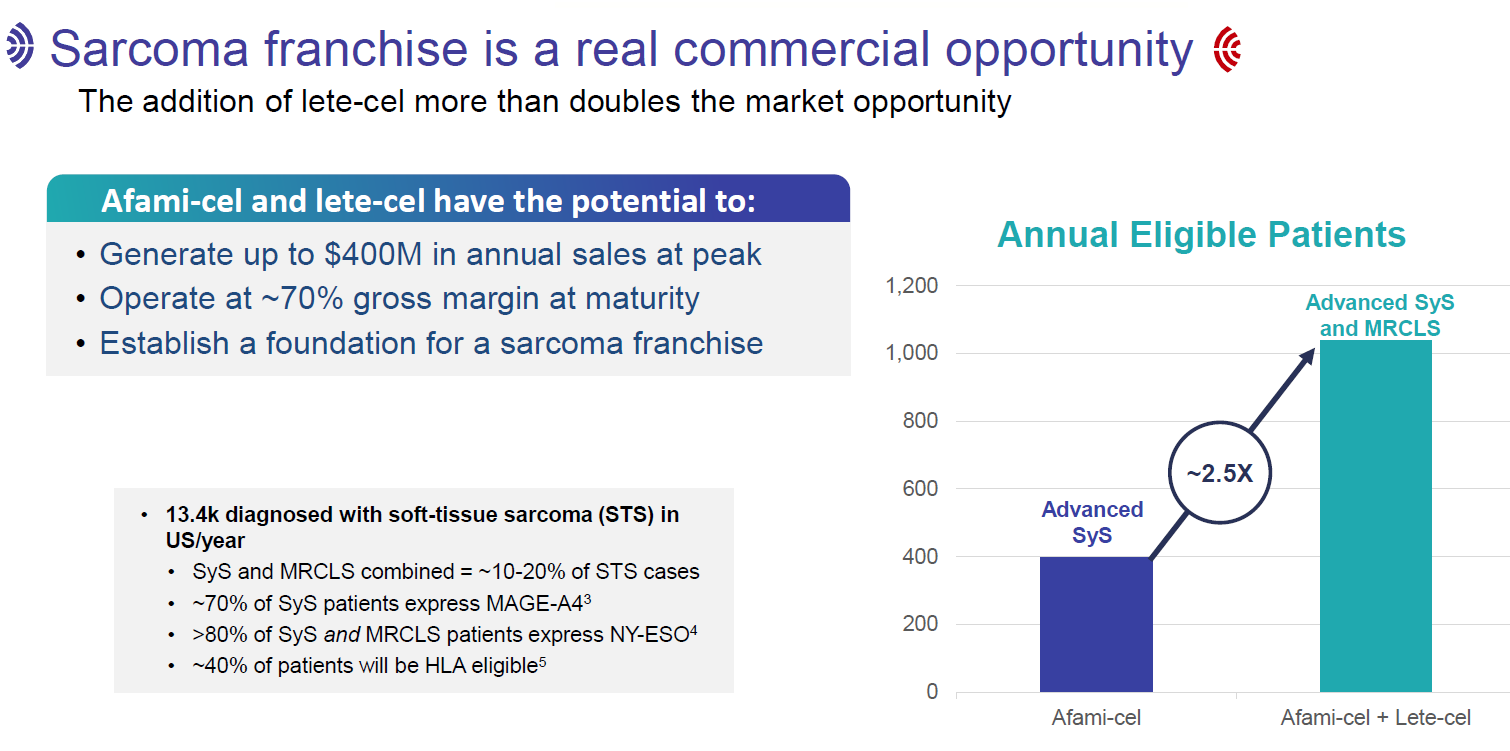
Adaptimmune预计Afami-cel和lete-cel合计将产生4亿美元的销售峰值。
图源:Adaptimmune官网
二、基于MDMA的创伤后应激障碍疗法
研发公司:Lykos Therapeutics
PDUFA日期:2024年8月11日
美国食品和药物管理局(FDA)正在审查3,4-亚甲基二氧基甲基苯丙胺(MDMA)结合心理干预辅助治疗创伤后应激障碍(PTSD)的NDA。如果获得批准,这将是首款MDMA辅助疗法,也是首款迷幻药辅助疗法。
该NDA得到了两项随机、双盲、安慰剂对照3期研究的支持:MAPP1(NCT03537014)和MAPP2(NCT04077437),MAPP1和MAPP2研究均达到了主要终点和次要终点,并发表在Nature Medicine杂志上。
这两项研究的结果表明,与安慰剂相比,MDMA辅助疗法能显著减轻创伤后应激障碍症状,其衡量标准是DSM-5创伤后应激障碍量表(CAPS-5)与基线相比的变化(主要终点)。此外,与安慰剂相比,MDMA还能明显改善与创伤后应激障碍相关的功能障碍,其衡量标准是Sheehan残疾量表(SDS)功能损害评分的变化(关键次要终点)。

MDMA辅助治疗试验疗效数据(图源:Lykos官网)
在最近的一次会议上,美国FDA精神药物咨询委员会以临床试验数据难以解释为由,投票反对批准该药物。委员会还对该药物的心脏和肝脏安全性以及缺乏有关其滥用可能性的数据表示担忧。
三、Seladelpar
研发公司:Adaptimmune Therapeutics
PDUFA日期:2024年8月14日
美国FDA已接受seladelpar用于治疗PBC患者的新药申请并授予其优先审评资格,其中包含用于治疗瘙痒症状以及那些对熊去氧胆酸(UDCA)应答不足或不耐受的非肝硬化或代偿性肝硬化的成人患者。FDA预计将于2024年8月14日前公布审评结果。今年2月12日,吉利德以43亿美元的价格收购CymaBay Therapeutics,将其主要候选产品为seladelpar收入囊中。
Seladelpar是一种在研的首类口服选择性过氧化物酶体增殖激活受体δ激动剂。Seladelpar的NDA申请获得了优先审评,并得到了多项试验数据的支持,包括3期RESPONSE(NCT04620733)和ENHANCE(NCT03602560)研究。这些试验招募了对熊去氧胆酸反应不充分或不耐受的原发性胆汁性胆管炎(PBC)患者(无肝硬化或有代偿性肝硬化)。

6月5日,吉利德公布了进行中ASSURE研究的两年中期结果。ASSURE研究中,参试者每天接受一次10 mg的Seladelpar治疗,疗程长达155 周。为期两年的中期分析的数据显示(包括来自参与过任何先前与seladelpar相关临床研究(传统研究)的179名受试者,和来自关键3期RESPONSE研究的158名受试者):
在完成24个月seladelpar治疗的99名来自传统研究的受试者中,70%达到了复合应答终点,包括碱性磷酸酶(ALP)水平低于正常上限(ULN)的1.67倍、ALP水平下降至少15%以及总胆红素(TB)水平等于或低于ULN。此外,其中42%的受试者在24个月时实现了ALP正常化,ALP是肝病进展的标志。对于来自传统研究的164名完成12个月seladelpar治疗的受试者,73%达到了具有临床意义的复合应答终点,42%的ALP恢复正常。在那些在随机接受seladelpar治疗后完成12个月RESPONSE研究,并继续接受总共18个月的持续seladelpar治疗(n=102)的受试者,62%达到了复合应答终点,33%达到了ALP正常化。
在整个ASSURE研究过程中,还使用数字评分量表(NRS;0-10)收集了患者报告的瘙痒情况。在基线NRS≥4的受试者中,观察到瘙痒持续改善。在ASSURE研究中,受试者在12个月和24个月时分别平均减少3.8分和3.1分。
四、Linvoseltamab
研发公司:Regeneron Pharmaceuticals
PDUFA日期:2024年8月22日
2024年02月,再生元(Regeneron Pharmaceuticals)宣布美国FDA已接受其BCMA/CD3双特异性抗体linvoseltamab的生物制品许可申请(BLA)并授予优先审评资格,用以治疗患有复发/难治性(R/R)多发性骨髓瘤(MM)的成年患者,患者在接受至少三种既往疗法后发生疾病进展。该申请的PDUFA日期为2024年8月22日。


Linvoseltamab是一款BCMA/CD3双特异性抗体,采用了再生元独有的全人源抗体技术VelocImmune和全长双抗技术平台VelociBi,与人体天然的抗体非常接近。该产品能够将表达CD3的T细胞与多发性骨髓瘤细胞上的B细胞成熟抗原(BCMA)桥接,以促进T细胞活化和对癌细胞的杀伤。
该上市申请主要基于一项I/II期LINKER-MM1研究(NCT03761108)数据的支持。LINKER-MM1是一项开放标签、多中心I/II期剂量递增和剂量扩展试验,旨在评估linvoseltamab在R/R MM患者中的疗效。去年12月公布的LINKER-MM1研究中期分析结果显示,在11个月的中位随访期间,117名接受剂量为200 mg的linvoseltamab治疗的患者中观察到客观缓解率为71%,46%达到完全缓解或更佳。
目前,全球共有两款BCMA/CD3双抗获批上市,分别为强生的Tecvayli和辉瑞的Elrexfio。若再生元的Linvoseltamab获批上市将成为全球第三款BCMA/CD3双抗。
五、Axatilimab
研发公司:Incyte&Syndax
PDUFA日期:2024年8月28日
2024年2月27日,Incyte宣布,美国FDA已经接受了抗CSF-1R抗体axatilimab的生物制品许可申请(BLA)的优先审查,该抗体用于治疗既往接受过二线及以上治疗后疾病进展的慢性移植物抗宿主病(GVHD)患者,指定PDUFA行动日期为2024年8月28日。
Axatilimab(SNDX-6352)是一款靶向CSF-1R的单克隆抗体,与CSF-1R具有高亲和力结合,并阻断两种已知的CSF-1R配体CSF-1和IL-34的结合。Axatilimab是根据Syndax和UCB于2016年签订的UCB全球独家许可开发的,2021年9月,Syndax和Incyte就Axatilimab签订了全球独家联合开发和联合商业化许可协议。
该申请得到了2期AGAVE-201研究(NCT04710576)数据的支持,该研究评估了axatilimab在241名患有复发性或难治性活动性GVHD的成人和儿童患者中的安全性和有效性,这些患者的疾病在接受2种治疗后出现进展。先前的治疗包括ruxolitinib(74%)、ibrutinib(31%)和belumosudil(23%)。
研究参与者被随机分配到3个治疗组之一:每2周0.3毫克/千克、每2周1毫克/千克或每4周3毫克/千克。主要终点是总缓解率(ORR),定义为每个剂量组中在第7周期第1天达到基于2014 NIH慢性GVHD共识标准的客观缓解的患者比例。

AGAVE-201研究试验设计(图源:Incyte官网)
研究结果显示,在治疗的前6个月内,每2周接受0.3mg/kg的患者的ORR最高,为74%(95% CI,63-83)。该队列中的中位反应时间为1.7个月(范围为0.9-8.1);这些患者中有60%在12个月时保持反应。在0.3mg/kg剂量组中,55%的患者在改良的Lee症状量表评分(次要终点)中至少获得了7分的改善。

AGAVE-201研究结果(图源:Incyte官网)
总结
2024年上半年,美国FDA批准了21款1类新药,其中包括17个化药和6个生物药。从适应症来看,罕见病为美国新药开发和获批的重点领域。抗肿瘤药物占比最高(29%),其次是抗感染(14%)、皮肤病(10%)、血液疾病(9%)、免疫系统疾病(9%)、心血管疾病用药(9%)。2024年下半年,相信会延续上半年的精彩,有更多具有里程碑意义的重磅药物获得批准。
参考资料:各公司公告
<END>
想要解锁更多药物研发信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!












 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论