
































开发仿制药的过程,往往就是对原研制剂深入理解的一个过程。要想对原研制剂的理解足够深入,就要对其底层的设计逻辑有所了解(《仿制药研发中的专利挖掘——制剂篇》)。
一般做仿制药的第一步,就是确定产品的QTPP,即Quality Target Product Profile(目标产品质量特性)。对于注射剂来说,我们会拉出这么一个表格:
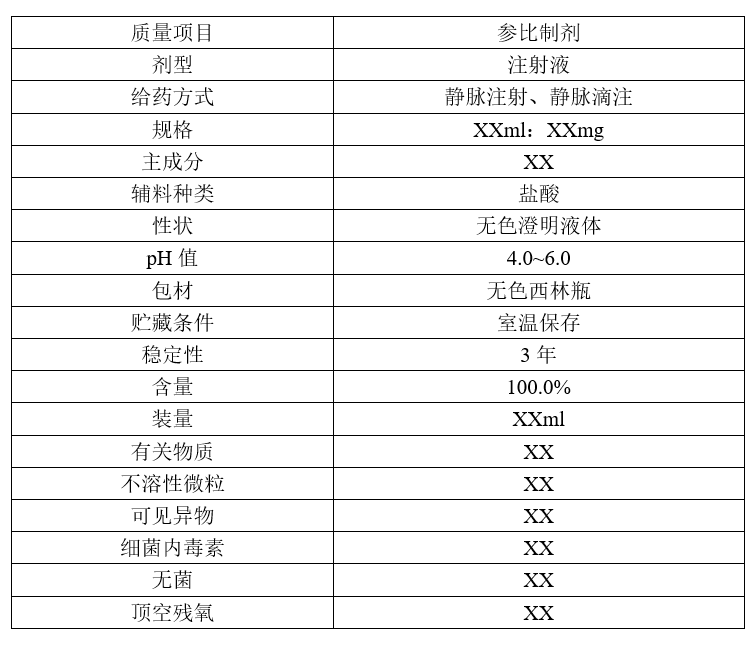
再从中选取关键质量属性(Critical Quality Attributes, CQA ),在处方和工艺研究中达到仿制药CQA和原研制剂一致。
如果仅仅是这样做,虽然大部分问题仿制药能够顺利开发出来,但有时还是会在困难的地方卡壳,更会陷入知其然不知其所以然的境地。为了更好地理解原研制剂的设计逻辑,需要把视野放得广一点,我们把QTPP去掉一个Q,就成了TPP,即Target Product Profile(目标产品特性)。TPP是做创新药的工具,是更大的概念,TPP描述了最终用户将如何使用产品。对企业而言,TPP将有助于确定项目目标和潜在风险。
TPP包含的范围很广,除了上面所说的产品质量特性外,还包括药物治疗相关的属性,法律相关属性及制造相关属性等等。药物治疗相关的属性,包含适应症、给药途径、剂量范围、给药频率、预期治疗时间、给药速率/时间、剂量、药物配伍等等;法律相关属性即是否侵犯专利;制造相关属性包括工艺是否和设备匹配、生产周期、销售成本等。药品的设计考虑到所有的这些方面,并恰当地予以解决,就是所谓的研发。例如,某种药物,为了达到最佳的治疗效果,需要设计成肌肉注射的水针剂。肌肉给药的话,给药剂量受到限制,过大的体积很难进行注射,一般限制在4ml以下。为了达到药理作用,需要在这不到4ml的体积里给到足够的活性物质,即需要保证API的可溶化和稳定性。而在使药液稳定的中性pH范围内,API的溶解性很低。这时设计添加合理的辅料,使API能够在中性条件下增溶,达到治疗作用的浓度,即成为研究的重点。
正因为一个药品的研发需要考虑到方方面面,所以并不是每个属性都能够达成最优解。这时,我们会将一些指标妥协、放宽,以将药品成功开发出来。这就涉及到一个“必须(must)”和“想要(wants)”的概念了。在开发过程中,人们通常会描述他们认为是产品“必须”的东西。例如,“产品必须等渗,pH值为5-8”或“必须有2年的保质期。”但上述这些说法其实并不恰当,提出的标准单一化且要求过高了。这里区分最低可接受要求(must)和期望属性(wants)是至关重要的。例如关于2年保质期,这可能是对许多注射剂产品的合理要求,以确保分销和储存期内的稳定性。然而,如果从公司的角度来看,相对于开发停止,推出一个只有18个月保质期的重磅产品更可取,那么2年的保质期就不是必须的。又如因为人体血液的pH值在7.4左右,药液的pH值控制在5-8范围内被认为在临床使用时是温和的。但其实很多的注射剂出于溶解度或是稳定性的考虑,pH值是在5-8范围外的。那么,“pH值为5-8”就不是“必须(must)”,而是“想要(wants)”。
对于注射剂质量属性中常见的“必须(must)”和“想要(wants)”,我们可以列一个表格说明。
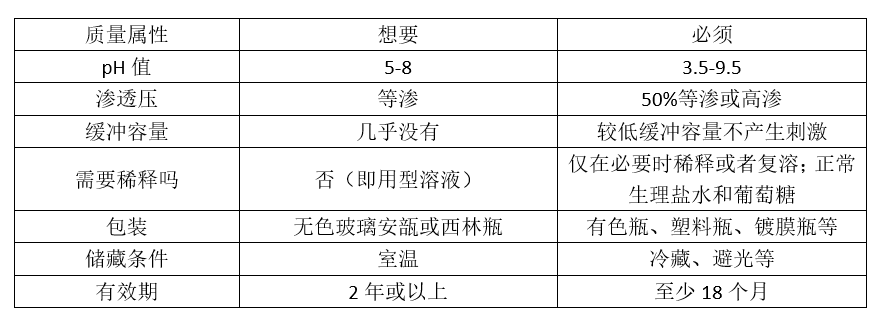
要提到的是,上述表格只是常规的情况,并不是绝对的。例如,不同给药途径的注射剂的要求是不尽相同的,比如静脉注射的注射剂需要达到等渗,而肌肉注射的注射剂需要达到一定的高渗,以促进临床给药时更好的吸收。又如,如果企业具有安瓿瓶灌装线,那安瓿瓶包装就是“想要”;而如果企业只有塑料瓶灌装线,那塑料瓶包装就是“想要”。
有了“必须(must)”和“想要(wants)”这种分级管理的思维方式,我们就可以采用建立标准化模型的方式理解原研制剂。例如,对于静脉给药的注射剂,可以认为如下处方工艺在临床使用上是最安全最理想的:
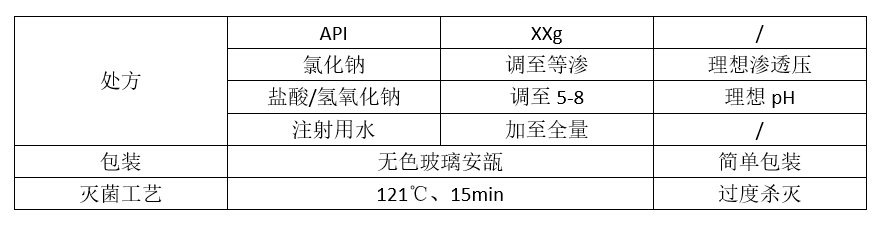
如果发现原研制剂与上述要求有不同的地方,那其往往是有不得已而为之。
举几个例子:
例1:盐酸莫西沙星氯化钠注射液

这个产品渗透压调至略低渗,是由于API的溶解度一般,而氯化钠又会和API产生同离子效应,因此要适当减少用量。储存时温度低于15℃会有析出,提升温度可复溶。原研在处方设计和储存条件上都选择了妥协的“必须(must)”方案。
至于pH值偏低,是由于pH调高后API的盐型会被破坏,溶解度也会降低。
例2:呋塞米注射液

在这个例子中,异常的是产品的pH值,调到了9左右,显然不是给药时对人体刺激最小的方案,这里原研制剂选择了妥协的“必须(must)”方案。带着问题调研文献,可以发现呋塞米在水中不溶,它需要和氢氧化钠成盐后溶解。而且原研制剂说明书提到药品低温时可能析晶,可40℃加热复溶后使用。由此看出,pH调至9时,呋塞米也只是刚刚可以溶解。这个产品设计时在临床适用性和药物溶解度两方面取得了平衡。理解了这一点,我们在做仿制药开发时,就会对原料药成盐的过程、制剂的可见异物和不溶性微粒进行重点关注和研究。
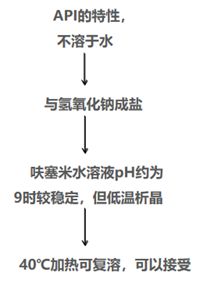
图 呋塞米注射液处方设计逻辑
从以上两个例子可以看到,原研制剂在临床的适用性和药学稳定性两方面发生矛盾时,会做出权衡,衡量风险收益比后在某个方面选择妥协的“必须(must)”方案。而有的品种,不同企业去做,权衡之后会选择不同的方案。
例3:地塞米松磷酸钠注射液
关于地塞米松磷酸钠注射液,国家局发布了两个参比制剂。
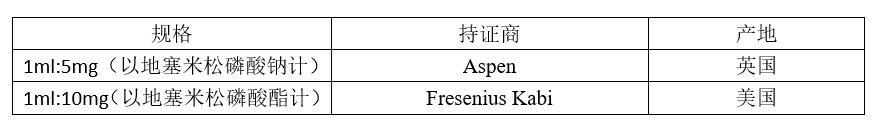
两个参比制剂的处方是这样的:

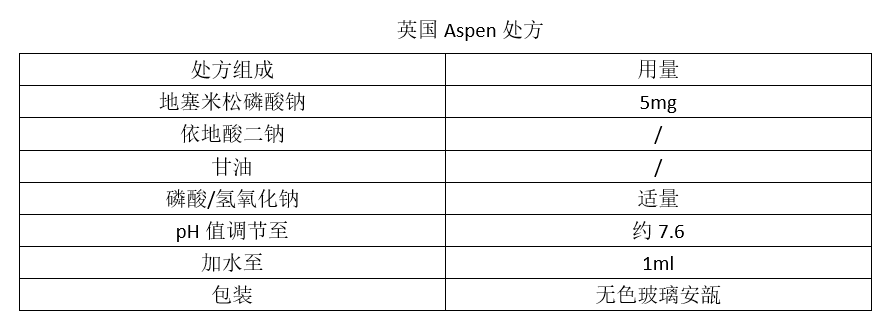
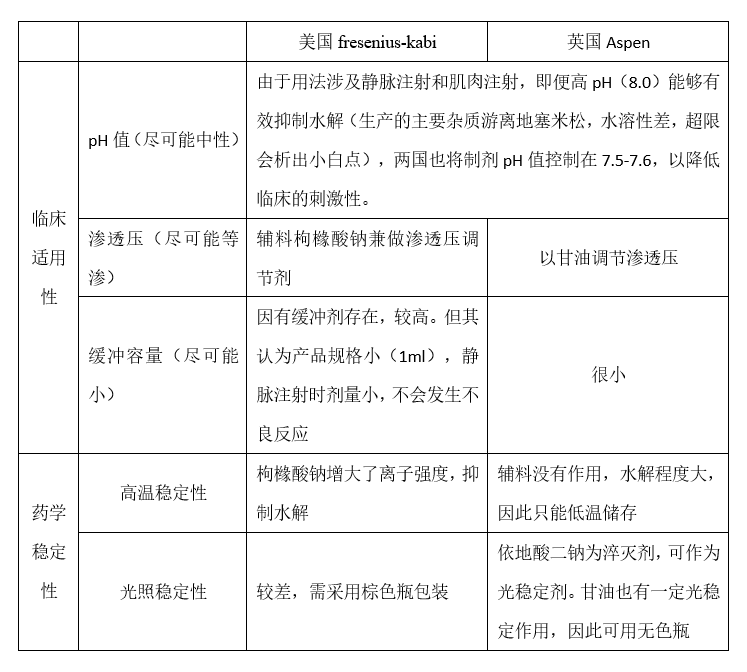
我们发现,两个参比制剂的处方截然不同,显示他们的设计思路也截然不同。如上所述,一个注射剂产品的设计,都要满足临床的适用性和药学稳定性两方面的要求。在这一点上,两个参比制剂是一样的。只是,他们走的方向不同。现用一张表格阐释两个参比制剂的设计思路。
另外一点值得注意的是,对于“必须”还是“想要”,不同国家地区的药监部门有着不同的要求。例如,对于无菌产品,欧盟严格执行灭菌决策树,首选终端灭菌方式;而日本则无此要求,企业可自主选择除菌方式。另外一个有趣的例子是维生素C注射液,下表是维生素C注射液在美国和日本上市的产品处方:
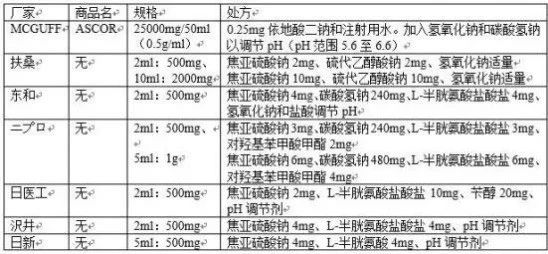
表格中第一行的ASCOR即是MCGUFF公司在FDA上市的维生素C注射液。与日本和中国早已上市应用数十年不同的是,ASCOR是FDA在2017年才首个批准的维生素C注射液。分析一下这个产品的信息可以发现,它的浓度比日本上市品高两倍以上,灌装量更是其十倍以上,处方里没有一般被认为这个产品必需的焦亚硫酸钠,储存温度是2-8℃,效期只有12个月(日本产品可室温保存2年)。说明书里规定其是在医院里由医护人员在4小时内稀释分装,为多个患者使用。按照前述“必须”还是“想要”的思路,我们可以合理推测出,FDA认为在这个营养类的注射剂中加入焦亚硫酸钠是不可接受的,风险收益比不理想。即维生素C注射液中不含亚硫酸盐是“必须”,而其他国家的药监部门认为只是“想要”。时至今日,终于有MCGUFF公司达成了FDA的要求,依托其强大的分销配送体系,让美国患者用上了更加安全的维生素C注射液。这样的产品如果被推选为参比制剂,我们真的可以仿制吗?技术上做出同样质量的产品可能并不困难,困难的是建立强大的分销配送体系,改变医生的用药习惯,让患者为用药安全承担更高的成本。
理解了产品的特性,理解了原研制剂的设计逻辑,理解了法规的制定思路,我们才能真正做到为患者着想,做出更加安全有效的注射剂。
欢迎关注药通社“注射剂系列大讲堂”

1)对注射剂原研制剂的深入剖析
2)谈谈注射剂的理化项目: pH值、渗透压、缓冲容量
3)注射剂的原辅料相容性试验
4)对无菌制剂中抗氧剂和抑菌剂的研究
5)注射剂中间体质量标准制定策略
6)灭菌决策树
7)美欧日中灭菌技术要求对比
8)灭菌设备性能优劣的判定
9)地塞米松磷酸钠注射液处方探讨
10)那些困难的注射剂项目,到底难在哪儿?
<END>

















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论