
































引言:渭城朝雨浥轻尘,客舍青青柳色新
一次正式的BE花费少则几十万多则数百万,消耗无数的人力物力,但在正式BE过程中,没有人能保证100%的成功。体外溶出完美的拟合经过严苛体内崩解、溶出、吸收、分布、代谢及排泄,相当一部分药物表现的并不完美,甚至出现BE不等效。
本文就如何提高BE通过率从BE实验设计、平均生物等效性(ABE)、参比制剂标度的平均生物等效性(RSABE )及实例解析这四部分进行深入分析,探寻BE的。
1、BE实验设计
1.1生物等效性研究决策树
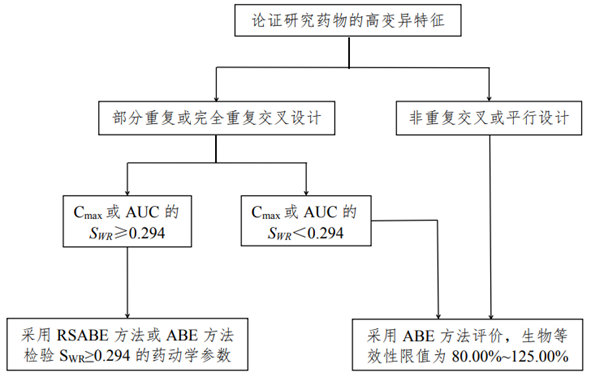
1.2试验设计
(1)交叉试验设计
①试验设计
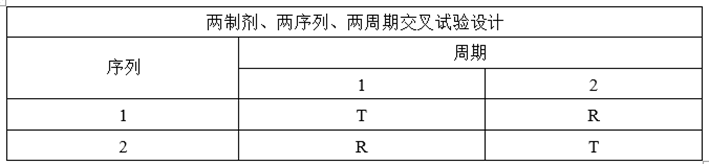
②适用药物
适用于一般药物,纳入健康志愿者参与研究,每位受试者依照随机顺序接受受试制剂和参比制剂。
③优点
1)交叉试验研究周期短,试验质量风险较低;
2)交叉试验研究周期短,脱落率较低;
④缺点
1)用于高变异药物,由于个体内变异较大,需要适当增加样本量,才可以满足试验的检验效能;
2)与重复试验相比,交叉试验得到的数据较少,统计学风险较高;
(2)平行试验设计
①试验设计

②适用药物
适用于半衰期较长的药物,每个制剂分别在具有相似人口学特征的两组受试者中进行试验。
③优点
1)单周期,消除半衰期较长药物的残留风险;
④缺点
1)与交叉试验相比,平行组设计需要更大的样本量;
2)需要相似人口学特征的受试者,受试者入组难度较大;
3)用于半衰期较长的药物,取样点设计难度较大;
(3)部分重复试验设计
①试验设计
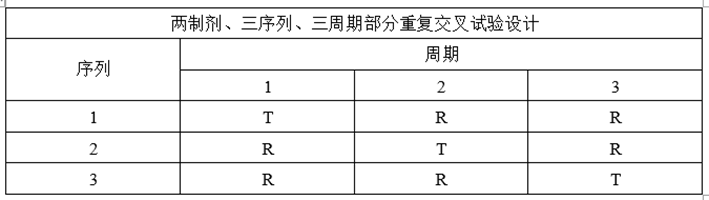
②适用药物
适用于部分高变异药物(个体内变异≥30%)。
③优点
1)可以入选较少数量的受试者进行试验,约为交叉试验设计受试者数量的3/4;
④缺点
1)部分重复交叉试验研究周期长,试验质量风险较高;
2)部分重复交叉试验研究周期长,脱落率较高;
(4)完全重复试验设计
①试验设计
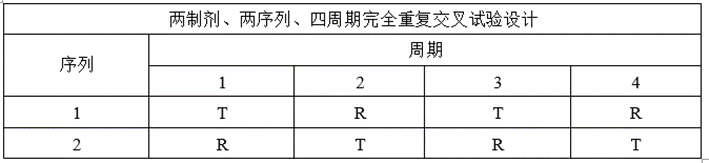
②适用药物
适用于部分高变异药物(个体内变异≥30%)。
③优点
1)可以入选较少数量的受试者进行试验,约为交叉试验设计受试者数量的1/2;
④缺点
1)完全重复交叉试验研究周期长,试验质量风险较高;
2)完全重复交叉试验研究周期长,脱落率较高;
2、平均生物等效性(ABE)
2.1 ABE方法简介
假设在正态分布下,试验制剂T与参比制剂R的生物利用度(BA)参数平均值相等。
仅比较两种制剂BA参数的边缘分布函数的均数或中位数,只能保证平均效应(均值)相同,不能保证其效应的变异度(方差)相同,但未考虑药物在体内代谢的差异,即个体内变异和个体与制剂间的交互作用,不能保证个体间的生物利用度相近,对低变异和高变异药物设置的等效性标准是一样的。
2.2 ABE接受标准
受试制剂与参比制剂的主要药动学参数(AUC 和 Cmax)几何均值比的 90%置信区间落在 80.00%~125.00%范围内。
2.3 ABE影响因素
(1)药物因素
①溶解性等原料药的理化性质;
②药物溶出等制剂的处方因素;
③药物分布、首过代谢、全身代谢和清除等药物固有性质;
(2)机体生理因素
①胃肠道 pH 值、胃肠动力、胃排空、小肠转运和结肠驻留时间等影响生物利用度的生理因素;
②饮食等其他因素;
2.4提高ABE策略
①开发出具有区分力的溶出参数;
②根据具有区分力的参比制剂溶出数据调整自制制剂处方工艺,提高自制制剂与参比制剂的体外溶出拟合度;
③加强临床试验管理,提高临床试验质量,降低由于受试者空腹试验偷吃零食、吐药、不正常脱落等风险;
④获得预BE数据,根据预BE数据调整自制制剂处方工艺,通过提高自制制剂与参比制剂的体外溶出拟合度进而提高自制制剂与参比制剂的体内拟合度;
⑤根据预BE数据调整试验方案,如增加样本量等措施,降低偶然性风险,利用计算机模型模拟正式BE,有的放矢的调整正式BE的试验方案,进而提高正式BE的通过率;
3、参比制剂标度的平均生物等效性(RSABE )
3.1高变异药物(HVD)
某些药物由于生物利用度过低、酸不稳定、吸收前的广泛代谢等原因,导致一个或多个药动学参数的个体内变异系数(Within-subject coefficient of variation, CVW%)大于或等于 30%,称为高变异药物(Highly variable drug, HVD)。
3.2 RSABE方法简介
对于安全性较好、治疗窗较宽的高变异药物,在充分科学论证的基础上和保证公众用药安全、有效的前提下,通过部分重复或完全重复交叉设计,根据参比制剂的个体内变异,采用参比制剂标度的平均生物等效性(Reference-scaled average bioequivalence, RSABE)方法,将等效性判定标准在80.00%~125.00%的基础上适当放宽,可减少不必要的人群暴露,达到科学评价不同制剂是否生物等效的目的。
3.3 RSABE接受标准
若的单侧 95%置信区间上限小于等于零,同时,制剂间主要药动学参数的几何均值比(Geometric mean ratio, GMR)的点估计值在 80.00%~125.00%范围内,可判定受试制剂与参比制剂的药动学评价指标(AUC 或 Cmax)具有生物等效性。只有 AUC 和 Cmax均判定等效才可申明该制剂与参比制剂具有生物等效性。
3.4 RSABE影响因素
(1)药物因素
①溶解性等原料药的理化性质;
②药物溶出等制剂的处方因素;
③药物分布、首过代谢、全身代谢和清除等药物固有性质;
(2)机体生理因素
①胃肠道 pH 值、胃肠动力、胃排空、小肠转运和结肠驻留时间等影响生物利用度的生理因素;
②饮食等其他因素;
3.5提高RSABE策略
在提高ABE策略的基础上
①试验通常应在同一中心完成;
②科学的试验设计,并应避免试验质量对个体内变异的估计引入偏倚;
③严格受试者入组条件,避免不合格受试者入组,增大不必要的个体内变异;
4、实例解析
4.1 ABE方法
(1)预BE实验设计
某仿制药XXX片,为薄膜包衣片,为低变异短半衰期品种,参比制剂资料显示个体内变异系数<0.3,该制剂餐后变异系数大于空腹,预BE选择餐后给药。
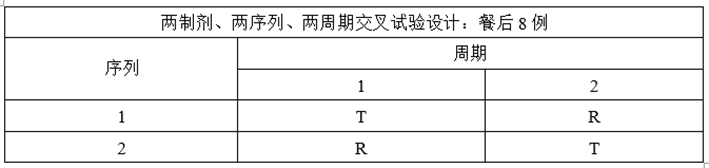
(2)溶出与预BE数据
①溶出数据
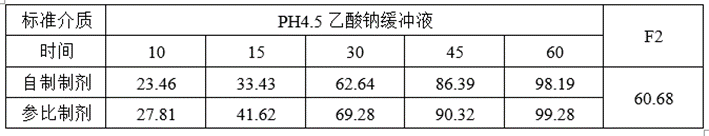
②预BE数据
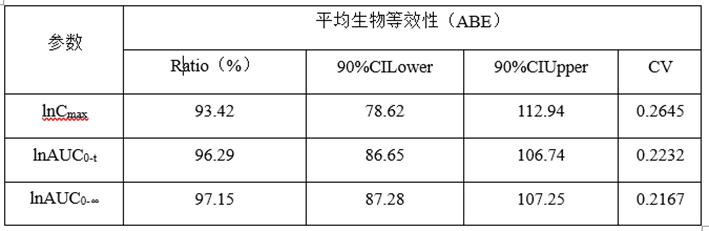
(3)预BE数据分析
①预BE数据分析
lnCmax的90%CILower78.62低于80%,lnCmax的T/R的Ratio为93.42%,自制制剂体外溶出慢于参比制剂,推测这可能是导致自制制剂体内Cmax低于参比制剂的主要原因。
lnCmax90%CI置信区间较宽(78.62%-112.94),可以通过增加正式BE样本量解决这一问题。
(4)完善ABE实验设计
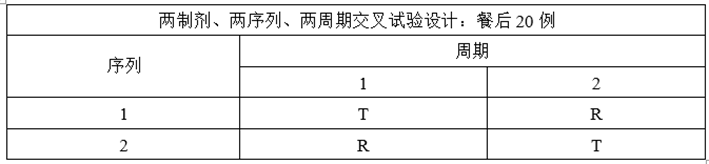
①调整自制制剂处方工艺,提高自制制剂体外溶出速率约5%;
②增加正式BE样本量至24例;
(5)ABE实验预分析
①溶出数据
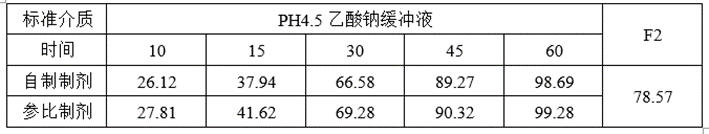
②计算机模拟ABE数据
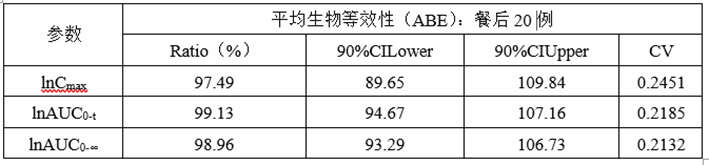
(6)正式ABE数据
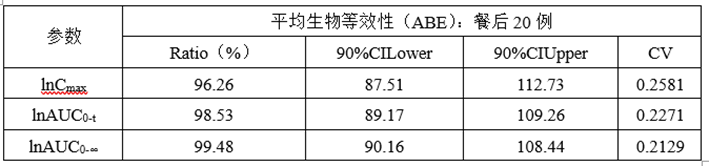
(7)总结
①研发前期需开发出具有区分力的溶出参数;
②根据具有区分力的参比制剂溶出数据调整自制制剂处方工艺,提高自制制剂与参比制剂的体外溶出拟合度;
③获得预BE数据,根据预BE数据调整自制制剂处方工艺,通过提高自制制剂与参比制剂的体外溶出拟合度进而提高自制制剂与参比制剂的体内拟合度;
④根据预BE数据调整试验方案,如增加样本量,降低偶然性风险,进而提高正式BE的通过率;
4.2 RSABE方法
(1)预BE实验设计
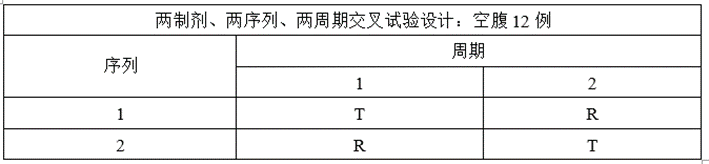
某仿制药XXX片,为薄膜包衣片,为高变异短半衰期品种,参比制剂资料显示个体内变异系数约为0.4,该制剂空腹变异系数大于餐后,预BE选择空腹给药。
(2)溶出与预BE数据
①溶出数据
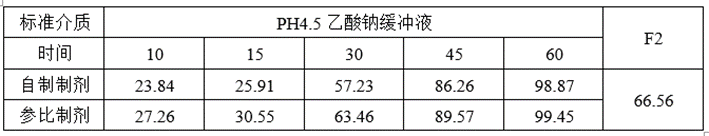
②预BE数据
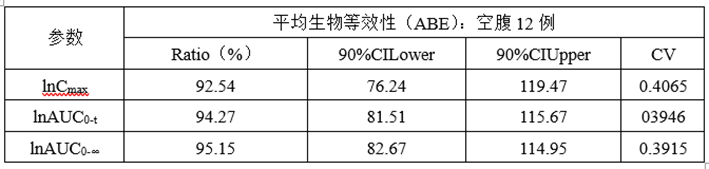
(3)预BE数据分析
①预BE数据分析
lnCmax的90%CILower76.24低于80%,lnCmax的T/R的Ratio为92.54%,CV为0.4065,为高变异品种,推测自制制剂体外溶出慢于参比制剂且个体内变异较高是导致自制制剂体与参比制剂不等效的主要原因。
(4)完善RSABE实验设计
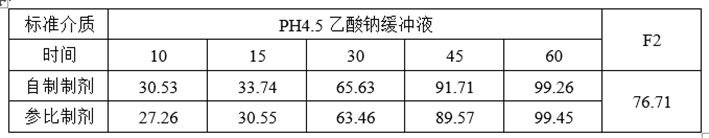
①调整自制制剂处方工艺,提高自制制剂体外溶出速率约6~10%;
②增加正式BE样本量至24例;
③将两制剂、两序列、两周期交叉试验设计调整为两制剂、三序列、三周期部分重复交叉试验设计;
④采用ABE法和RSABE法同步计算;
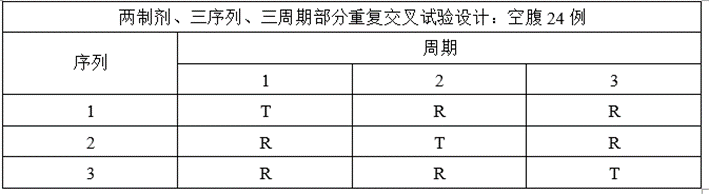
(5)ABE、RSABE实验预分析
①溶出数据
②计算机模拟ABE数据
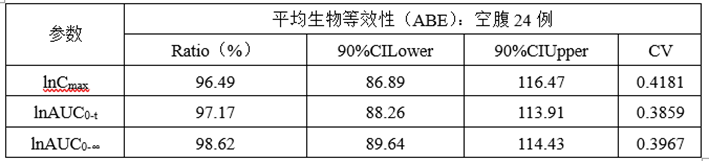
③计算机模拟RSABE数据
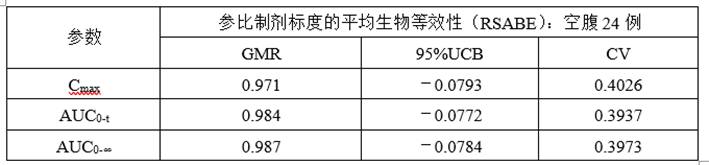
(6)正式ABE、RSABE实验
①正式ABE数据
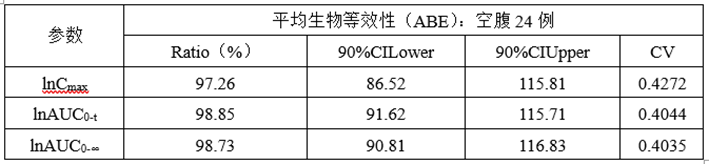
②正式RSABE数据
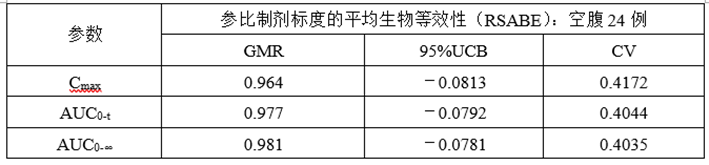
(7)总结
①研发前期需开发出具有区分力的溶出参数;
②根据具有区分力的参比制剂溶出数据调整自制制剂处方工艺,提高自制制剂与参比制剂的体外溶出拟合度;
③获得预BE数据,根据预BE数据调整自制制剂处方工艺,通过提高自制制剂与参比制剂的体外溶出拟合度进而提高自制制剂与参比制剂的体内拟合度;
④根据预BE变异率调整试验方案,将两制剂、两序列、两周期交叉试验设计调整为两制剂、三序列、三周期部分重复交叉试验设计,方便后续采用RSABE评价法;
⑤根据预BE数据调整试验方案,如增加样本量,降低偶然性风险,进而提高正式BE的通过率;
⑥严格受试者入组条件,避免不合格受试者入组,增大不必要的个体内变异;
⑦采用ABE法和RSABE法同步计算,最终根据药物的安全性、治疗范围科学合理选择等效性评价方法;
5、总结:劝君更尽一杯酒,西出阳关无故人
如何抓住BE,这只薛定谔的猫?这需要从药物制剂的理化性质和胃肠道系统的生理状态这两方面因素来考量,在保证自制制剂和参比制剂理化性质一致的基础上,进行预BE试验,根据预BE试验结果,有的放矢的提高自制制剂与参比制剂的体外溶出拟合度,调整正式BE的试验设计,利用计算机数据模型进行模拟,根据计算机数据模型模拟的BE数据科学合理的调整正式BE的试验设计,降低假阳性和假阴性的风险,进而提高BE的通过率。
声明:作者水平有限,不可避免有错误或不及时的信息,欢迎留言指出。
参考文献:
1.国家食品药品监督管理总局《普通口服固体制剂溶出曲线测定与比较指导原则》(2016 年 3 月).
2.国家食品药品监督管理总局《普通口服固体制剂溶出度试验技术指导原则》(2015 年 2 月).
3.国家食品药品监督管理总局《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》(2016 年 3 月).
4.国家食品药品监督管理总局《人体生物等效性试验豁免指导原则》(2016 年 5 月).
5.国家食品药品监督管理总局《已上市化学药品药学变更研究技术指导原则(试行)》溶出曲线研究的问答(2022 年11 月).
6.国家食品药品监督管理总局《药物临床试验的生物统计学指导原则》. 2016 年 6月.
7.国家食品药品监督管理总局《高变异药物生物等效性研究技术指导原则》. 2018 年10月.
<END>
要解锁更多企业药品研发信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药品各国上市情况、药品批文信息、销售情况与各维度分析、市场竞争格局、一致性评价情况、集采中标情况、药企申报审批信息、最新动态与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论