




























近年来,罕见病是药企乃至整个社会都很关心的话题。
罕见病通常指发病率极低的疾病,又称“孤儿病”。世界各国根据各自实际情况,在罕见病的定义上略有不同。美国将罕见病定义为患者少于20万人/年或其治疗药物销售额预期 难以收回研发成本的疾病;欧盟将患病率小于0.5‰、能导致衰弱或威胁生命的疾病定义为 罕见病;日本将患者少于 5万/年或患病率小于0.5‰作为罕见病定义标准。我国在2021年《中国罕见病定义研究 报告 2021》中将罕见病定义为“新生儿发病率小于1/万、患病率小于1/万且患病人数小于14万的疾病”。
其中,80%的罕见病是由遗传缺陷或基因突变造成的,而细胞与基因疗法也正成为罕见病药物研发的热点:相对于常见疾病,罕见病通常由单个基因突变引起,是基因治疗的理想靶标;约有超过90%的罕见病目前缺乏有效治疗药物,基因和细胞治疗作为一种潜在的创新方法,有望为罕见病治疗带来突破性改善。细胞与基因疗法在治疗罕见病方面已展现出重大进展,有望为全球2.6-4.5亿受罕见病影响的人群(包括儿童)带来治愈希望。
今天药融圈要介绍的这家公司叫Ultragenyx,它的产品和研发管线均是罕见病药物。

Ultragenyx Pharmaceutical(纳斯达克股票代码:RARE)于2010年在硅谷成立,是一家专注于罕见病药物研发与商业化美国生物技术公司。经过2轮融资后,于2014年成功在纳斯达克上市。(截至2024年3月8日,该公司股价50.53美元/股,市值约41.6亿美元。3月26日,市值为37.43亿美元。)其创始人、总裁兼首席执行官Emil Kakkis曾在Harbour-UCLA医疗中心工作,并开发一种针对罕见疾病MPS I 的酶替代疗法。
目前,Ultragenyx公司拥有一支在罕见病治疗开发和商业化方面经验丰富的管理团队,在团队的努力下,公司已有4款产品上市,且建立起了一个多元化的候选疗法管线,旨在解决未被满足的医疗需求。其中,Crysvita被批准用于治疗X连锁低磷血症(XLH),以及肿瘤性低磷骨软化症(TIO)。
管线布局&产品
Ultragenyx目前批准的疗法和临床阶段的产品线包括四个产品类别:生物制剂、小分子、AAV基因疗法和核酸候选药物,有四种已获商业上市批准的产品:用于治疗X-连锁低磷血症(XLH)和肿瘤诱导的骨软化症(TiO)的Crysvita®(burosumab)、用于儿童和成年人黏多糖贮积症VII型(MPSVII)或SLY综合征的Mepsevii®(vestronidase Alfa)、用于治疗长链脂肪酸氧化障碍(LC-FAOD)的Dojolvi®(triheptanoin)和用于治疗家族性高胆固醇血症(HoFH)的Evkeeza®(evinacumab)。

上市产品
1.Crysvita®(burosumab/布罗索尤单抗)
CRYSVITA于2018年4月在美国获批上市。CRYSVITA是成纤维细胞生长因子23 (fibroblast growth factor23, FGF23)的阻断抗体。当时获批的适应症是6个月以上的儿童及成人X连锁低磷酸盐血症(X-linked hypophosphatemia, XLH, 又名:家族性低磷酸血症佝偻病)。
XLH是一种罕见的、遗传性的、进行性的和终生的骨骼肌肉系统疾病。其病因是体内调磷因子FGF23产生过多或降解障碍,使循环中FGF23水平增加,导致肾脏排磷增多及低磷血症。估计所有的发达国家中有约48,000个病人,包含约36,000成人和12,000儿童。
在2020年6月,FDA批准了CRYSVITA的第2个适应症:2岁及以上的儿童及成人肿瘤性骨软化症的低磷酸盐血症(FGF23相关)。
肿瘤性骨软化症通常是由产生过量FGF23的良性肿瘤所引起的,该病将会导致严重的低磷酸盐血症、骨折以及肌无力。一般首选的治疗方案是手术切除肿瘤,但对于某些患者,肿瘤无法切除或肿瘤切除后复发。对于这些患者仍然存在着巨大的未满足临床需求。估计所有的发达国家肿瘤性骨软化症的患者数量约为2,000-4,000。
除了在美国,目前CRYSVITA还在加拿大,欧洲地区(欧盟、英国、瑞士等),南美, 土耳其等地获批。Ultragenyx正在与日本巨头协和麒麟/Kyowa Kirin Co.,Ltd.(简称KKC)以及KKC的全资子公司Kyowa Kirin合作,在全球范围内开发和商业化Crysvita。中国也已经获批上市。

2.Mepsevii®(vestronidase Alfa)
MEPSEVII于2017年11月在美国获批上市。该药是一种重组人溶酶体β葡萄糖醛酸酶(酶替代疗法),用于儿童和成年人黏多糖贮积症VII型(MPS VII,斯莱综合征【Sly syndrome】)的治疗。
黏多糖贮积症(mucopolysaccharidosis,MPS)是一组复杂的、进行性多系统受累的溶酶体病,是由于降解糖胺聚糖的酶缺乏所致。黏多糖贮积症共分为7型。黏多糖贮积症VII型是最罕见的溶酶体贮积症亚型之一,估计所有的发达国家中只有约200个病人,国内目前仍然缺乏大样本流行病学数据。VII型通常会导致多器官功能衰竭,广泛性骨骼功能障碍和死亡。
除了在美国,目前MEPSEVII还在欧盟、英国,巴西和墨西哥获批。
3.Dojolvi®(triheptanoin)
Dojolvi于2020年6月在美国获批上市。该药是一种高纯度的、合成的7碳脂肪酸甘油三酯。它是一种中链、奇数碳脂肪酸,可作为能量来源及体内各反应的代谢产物的替代品,用来治疗儿童和成人长链脂肪酸氧化障碍(Long-chain fatty acid oxidation disorders, LC-FAOD) 。
LC-FAOD是一组罕见的代谢性疾病,该病会抑制脂肪代谢,阻止脂肪转化为能量,并可能导致低血糖、肌肉撕裂、心脏以及肝脏疾病。估计所有的发达国家中有约8,000-14,000个病人。
4.Evkeeza®(evinacumab)
药融云数据显示:Evkeeza是一种通过静脉注射给药的全人单克隆抗体,可结合并阻断血管生成素样蛋白3(ANGPTL3)的功能,ANGPTL3是一种在脂质代谢中起关键作用的蛋白质,用于治疗纯合子家族性高胆固醇血症(HoFH)。Evinacumab是Evkeeza中的活性成分,它能结合到体内的一种名为ANGPTL3的蛋白,并阻断其作用。ANGPTL3涉及控制胆固醇水平,阻断其作用可降低血液中的胆固醇水平。Evkeeza通过每月(4周)一次的输注给药。本品为与再生元联合开发。
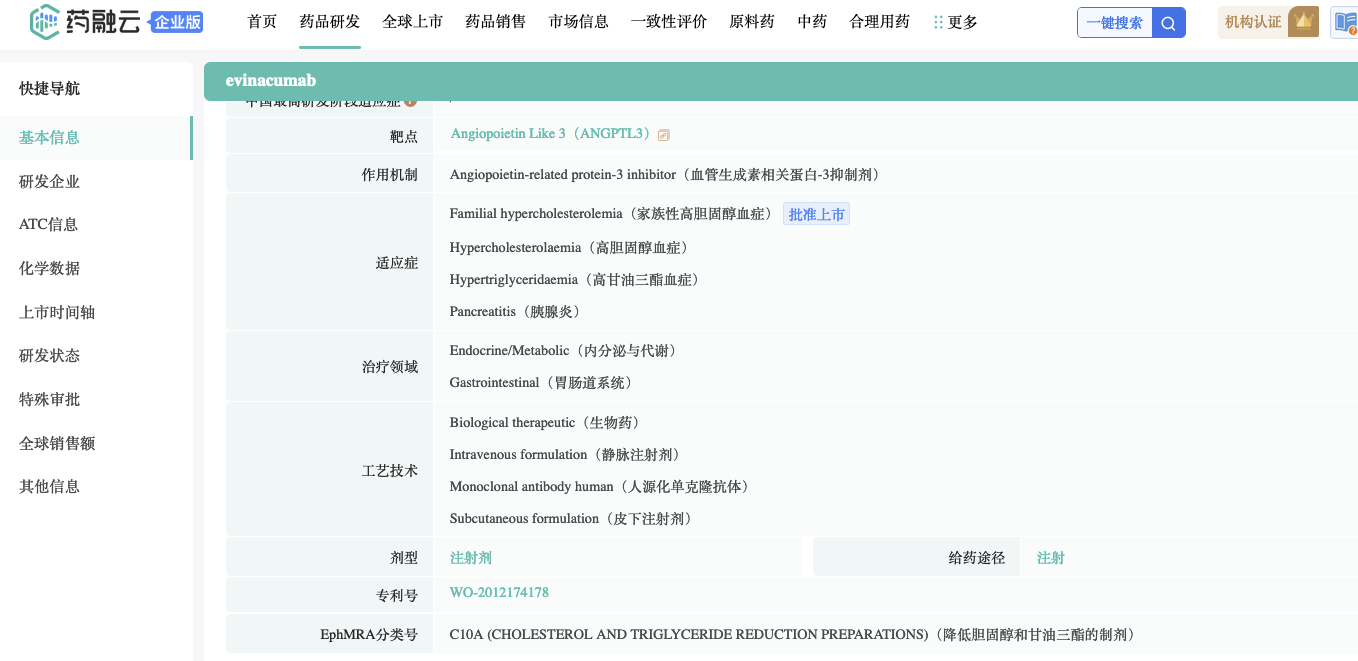
药融云数据,www.pharnexcloud.com
HoFH是一种极为罕见的遗传性疾病,是家族性高胆固醇血症(FH)中最严重的一种。Evkeeza是美国和欧盟批准的首个ANGPTI-3靶向疗法,用于辅助治疗12岁及以上HoFH患者。目前,Evkeeza正被评估用于5-11岁HoFH患者。在美国以外的发达国家,大约有3,000到5,000名HoFH患者。据估计,每100万人中约有4人患有HoFH,而且由于患者稀少,Evkeeza的治疗成本巨大,平均每年约45万美元。
管线更新和临床里程碑
1. UX143(Setrusumab)单克隆抗体治疗成骨不全症(OI):
ORBIT研究的3期部分预计将于2024年第一季度末全部纳入在后期临床试验Orbit和Cosmic中,患者正在接受给药,这些试验正在评估setrusumab在儿童和年轻成人OI患者中的应用。ORBIT研究的随机、安慰剂对照3期部分预计将招募约150名患者,并于2024年第一季度末全部招募。
ORBIT研究的其他长期2期安全性和有效性数据预计将于2024年下半年公布。3期COSMIC研究是一项活性对照研究,旨在评估setrusumab与静脉注射双膦酸盐(IV-BP)治疗相比,对2至5岁以下患者的年度总骨折率的影响。Cosmic预计将在全球20多个地点招募约50名或更多患者,预计将于2024年上半年完成招募。
2. GTX-102反义寡核苷酸治疗Angelman综合征:
1/2期完全入组;预计2024年上半年的扩张数据 扩大队列的招募工作于2023年12月完成,共招募了53名新患者。共有74名患者参加了1/2期研究,包括剂量递增/扩展研究患者。扩展队列将评估许多与先前纳入的剂量递增/扩展队列相同的安全性、药代动力学和疗效指标,以及一些新的评估。下一次安全性和有效性更新预计将于2024年上半年进行,并计划纳入至少20名扩展队列患者的数据,其中至少有170天的数据。
2024年1月,GTX-102被欧洲药品管理局(EMA)纳入优先药品(PRIME)计划。PRIME由欧洲药品管理局(EMA)授予那些根据早期临床数据显示有潜力使需求未得到满足的患者受益的药物。通过PRIME,EMA提供早期和主动支持,以优化开发计划,生成有关药物效益和风险的可靠数据,并加快对药物应用的评估。
3. UX701 AAV基因治疗Wilson病:
队列3中最后一名患者给药;预计中期第一阶段数据将于2024年中期发布 第1阶段的三个剂量递增队列中的所有患者均已给药。在第1阶段,将评估UX701的安全性和有效性,并在第2阶段(该研究的关键、随机、安慰剂对照阶段)选择剂量进行进一步评估。第一阶段的数据预计将在2024年中期公布,随后将在2024年下半年进行剂量选择并启动第二阶段。
4. UX111 AAV基因治疗Sanfilippo综合征(MPS IIIA):
2024年2月初,该公司在第20届年度世界研讨会(WorldSymposium™)上公布了一项正在进行的关键转移(Pivotal Transfer)研究的新的积极数据,该研究评估了UX111在患有MPS IIIA的儿童中的安全性和有效性,显示单次输注UX111可以显著纠正潜在的代谢疾病,并维持几乎所有患者的认知功能。该报告还显示,观察到的脑脊液中硫酸乙酰肝素暴露的减少可以预测UX111治疗后MPS IIIA患者的长期认知功能的改善。有了这些数据和其他数据,正在与FDA进行讨论,以寻求UX111的加速审查路径。
5. DTX401 AAV基因治疗Ia型糖原贮积病(GSDIA):
3期研究完成给药;第3阶段数据读出预计在2024年上半年。2023年5月,Ultragenyx宣布最后一名患者在3期研究中接受了给药。这项为期48周的研究纳入了8岁及以上的患者,按1:1的比例随机分配至DTX401或安慰剂组。3期安全性和有效性数据预计将于2024年上半年公布。
DTX301 AAV基因治疗鸟氨酸氨甲酰转移酶(OTC)缺乏症:3期研究给药患者;预计招生工作将于2024年上半年完成 在正在进行的3期研究中,Ultragenyx正在对患者进行随机分组和给药。关键的64周研究将包括约50名患者,按1:1随机分配至DTX301或安慰剂组。主要终点是通过去除氨清除剂药物和蛋白质限制饮食以及24小时氨水平的变化来测量的反应。招募工作预计将于2024年上半年完成。
财务状况
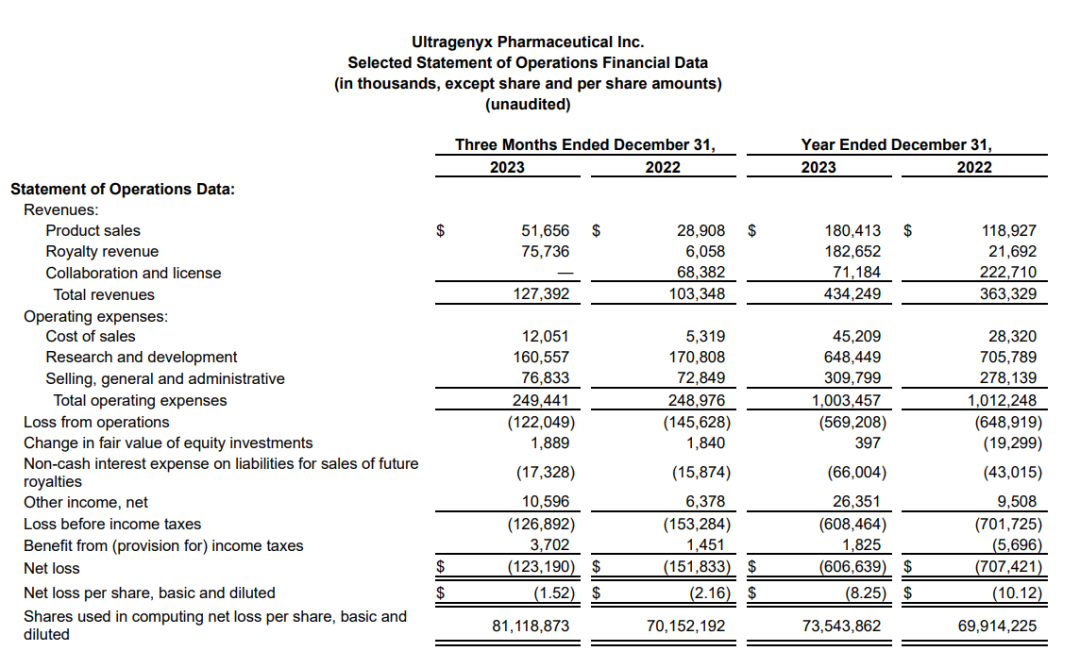
2024年2月22日,Ultragenyx公布2023财年年报。2023年Ultragenyx总营收为4.34亿美元,其中Crysvita收入3.25 ~ 3.3 亿美元;预计公司2024年总营收达到5.3亿美元。截至2023年末,该公司现金、现金等价物和有价债务证券约7.76亿美元。
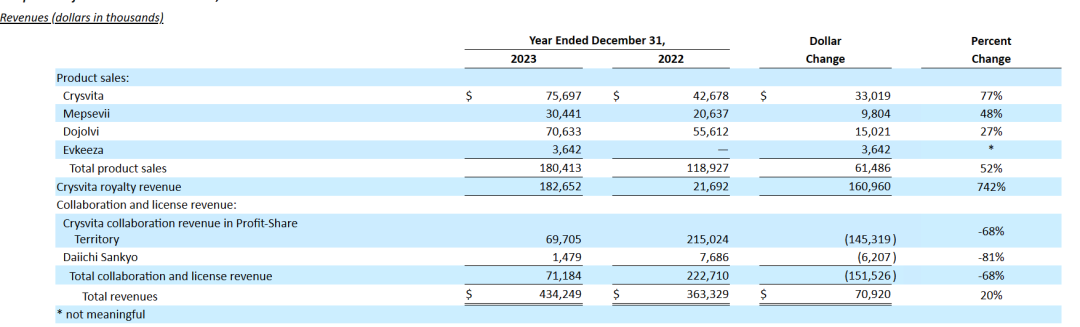
与2022财年相比,2023财年Ultragenyx的产品销售额增加了6150万美元。这一增长主要是由于拉丁美洲对Crysvita的需求增加,原因是接受治疗的患者数量增加,对其他批准产品的需求持续增加,以及指定患者计划的收入增加;Crysvita合作收入和特许权收入净增加了1560万美元。Crysvita收入的增加主要是由于接受治疗的患者数量增加;从Daiichi Sankyo/第一三共中获得的收入减少了620万美元,减少原因是2023年完成了技术转让和技术转让期。
下表是按主要项目类型和业务活动划分的研发费用明细(千美元):
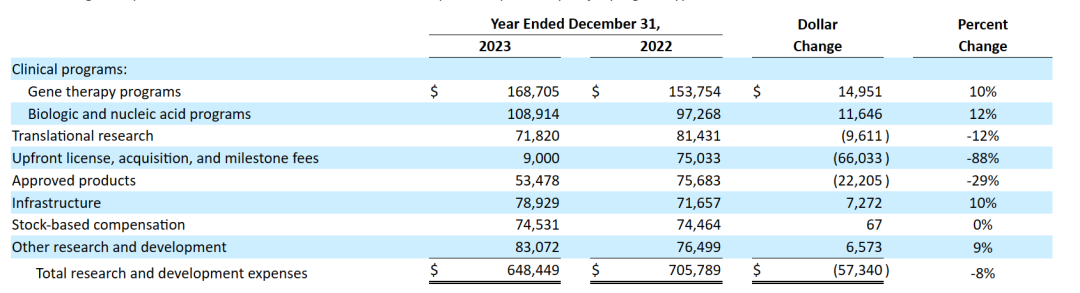
与2022财年相比,2023财年Ultragenyx的研发费用总额减少了5730万美元,主要由于:
• 对于基因治疗项目,增加1500万美元,主要与2022年5月从Abeona Therapeutics收购的UX111项目的开发成本增加以及内部制造的生产材料采购增加有关,部分被DTX401的外部临床制造费用的减少所抵消;
• 对于生物和核酸项目,增加1160万美元,主要与UX143和GTX102项目的持续临床进展以及相关的临床开发和制造费用有关,部分被用于治疗III型糖原贮积病的UX053开发费用的减少所抵消;
• 对于转化研究,减少了960万美元,主要与IND阶段项目的制造和员工人数费用减少有关;
• 前期许可、收购和里程碑费用减少6600万美元,原因是截至2023年12月31日确认的UX143项目临床登记里程碑的费用为900万美元,而截至2022年12月31日收购Genetx的费用为7500万美元;
• 对于批准的产品,减少2220万美元,主要是由于北美的Crysvita商业化责任转移到KKC,以及Dojolvi实现了成本效益,部分抵消了Regeneron协作费用的报销以及Evkeeza人员和产品发布成本的增加;
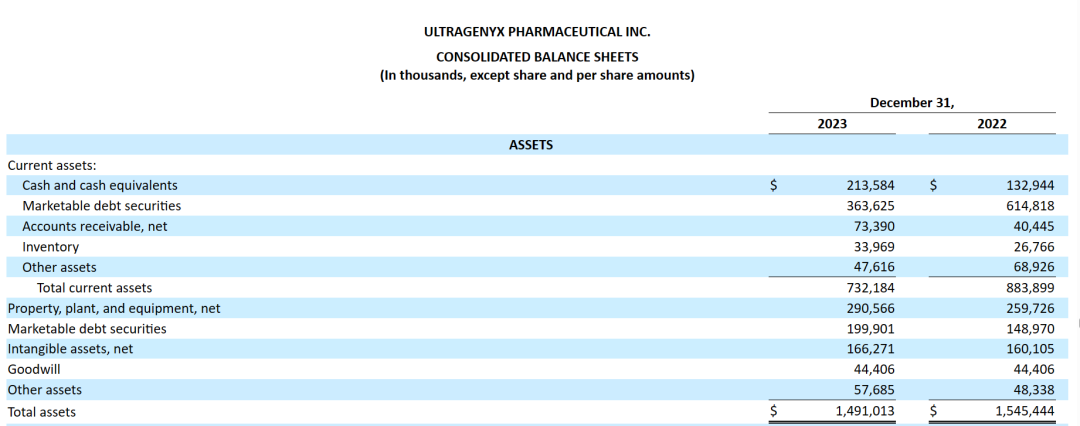
截至2023年12月31日,Ultragenyx拥有7.771亿美元的可用现金、现金等价物和可出售的债务证券,该公司称其现有资产将足以满足至少未来12个月的运营需求。
<END>
要解锁更多企业药品研发信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药品各国上市情况、药品批文信息、销售情况与各维度分析、市场竞争格局、一致性评价情况、集采中标情况、药企申报审批信息、最新动态与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!











 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论