



























据药融云统计,2022年美国FDA的CDER(药品评价与研究中心)共批准了37款新药上市,包括22款新分子实体和15款生物制品(《2022年美国FDA批准新药上市的37款药物盘点(一)》、《2022年美国FDA批准新药上市的37款药物盘点(二)》)。此外,美国FDA的CBER(生物制品评价和研究中心)还批准了8款新药上市,涉及4款基因疗法、1款细胞疗法、2款疫苗,以及1款微生物组疗法。CDER和CBER一共批准45款创新疗法,这也是美国FDA自2017年以来批准新药上市数量最少的一年。
2022年美国FDA的CBER批准的新药上市盘点
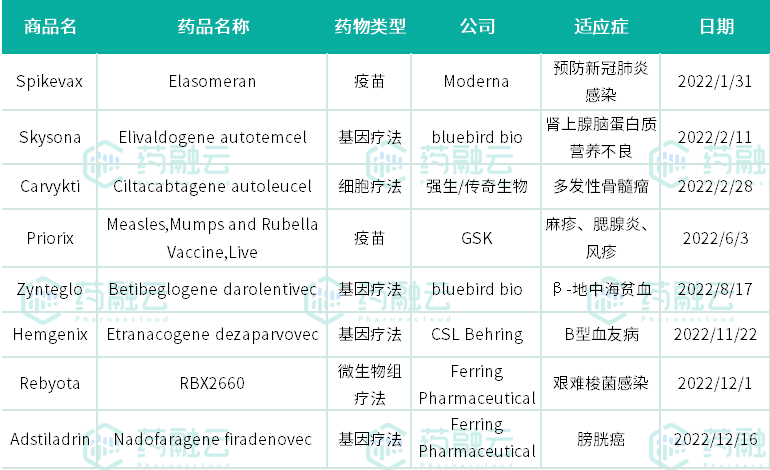
数据来源:药融云企业版数据库综合查询
资源下载
搜索并关注“药融云公众号(yrydata)”,后台回复关键词“FDA2022” ,即可下载《2022年FDA批准的45款新药和疗法》表格。
一、4款基因疗法,丰收的2022
2022年美国FDA批准的基因疗法是数量最多的一年,高达4款,其中罕见病治疗药物有3款。基因疗法定价普遍非常的高,2022年批准的这4款基因疗法也不例外。蓝鸟生物的Zynteglo在美国定价280万美元,Skysona定价300万美元,CSL Behring的Hemgenix定价更是达到了350万美元,成为全球最贵的药物。
- 01.Skysona:第一个治疗脑肾上腺脑白质营养不良(CALD)的基因疗法!
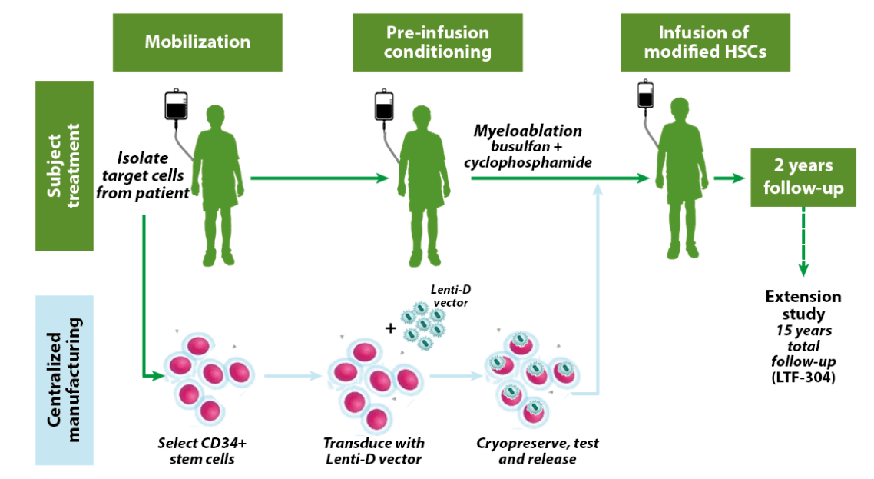
2022年2月11日,美国FDA批准Skysona(elivaldogene autotemcel,Lenti-D)的生物制品许可上市申请(BLA):该药是一种一次性基因疗法,用于治疗年龄在18岁以下的脑肾上腺脑白质营养不良(CALD)患者。CALD是一种罕见的神经退行性疾病,主要影响年幼儿童,可迅速导致进行性、不可逆转的神经功能丧失和死亡。
Skysona是第一个也是唯一一个获监管批准用于治疗CALD的一次性基因疗法。2021年7月,Skysona获得欧盟批准,用于治疗携带ABCD1基因突变、没有HLA匹配的同胞造血干细胞(HSC)供体可用的早期CALD患者。

CALD是一种进行性和不可逆的神经退行性疾病,它涉及髓鞘的破坏,髓鞘是大脑中负责思考和肌肉控制的神经细胞的保护鞘。CALD的症状通常发生在儿童期(中位年龄7岁),患者认知功能和身体功能快速渐进性下降。早期诊断CALD非常重要,因为治疗结局随疾病的临床阶段而变化。因此,必须在疾病快速进展之前进行治疗。若不进行治疗,近一半的患者会在症状出现起5年内死亡。Skysona的获批有着非常大的意义,给这一罕见病带了新的治疗选择。
此次批准基于II/III期Starbeam临床研究(ALD-102)(N=32)的功效和安全性数据支持,此外,BLA还包含III期临床试验ALD-104中35名受试者的数据。在ALD-102中,90%(29/32)的患者在24个月时达到主要评估终点,即无严重功能障碍(Major Functional Disabilities-free)的状态下生存。
根据相关数据,美国每年约有40~50名患者被诊断出患有CALD,蓝鸟生物预计每年约10名患者会使用Skysona进行治疗,市场潜力仍可进一步挖掘。
- 02.Zynteglo:首次批准慢病毒载体基因疗法
2022年8月17日,美国FDA批准Zynteglo(beti-cell)上市,是首个用于治疗需要接受常规血红细胞输注的β-地中海贫血患者的一次性基因疗法,适用于β-地中海贫血疾病的创新药物。Zynteglo是一种一次性基因治疗药物,单剂量给药,每个剂量的Zynteglo都是使用患者自身细胞(骨髓干细胞)创建的定制治疗,这些细胞经过基因改造以产生功能性β-珠蛋白,从而治疗β-地中海贫血疾病。

ZYNTEGLO的批准是基于HGB-207(Northstar-2)和HGB-212(Northstar-3)研究以及长期随访研究LTF-303的数据。
试验数据: ZYNTEGLO单臂、开放标签、为期24个月的3期研究招募了41名4-34岁的非β0/β0和β0/β0基因型患者,最长随访时间为4年。在不同年龄和基因型的可评估患者中,89%(32/36)实现了输血独立性,即至少12个月内不再需要红细胞输血,同时可维持平均总血红蛋白水平不少于9 g/dL。 (数据截止到最后一次随访)最常见的非实验室不良反应(≥20%)包括黏膜炎、发热性中性粒细胞减少、呕吐、发热、脱发、鼻出血、腹痛、肌肉骨骼疼痛、咳嗽、头痛、腹泻、皮疹、便秘、恶心、食欲下降、色素沉着障碍和瘙痒。最常见的3或4级实验室异常(>50%)包括中性粒细胞减少、血小板减少、白细胞减少、贫血和淋巴细胞减少。所有参与Northstar-2(HGB-207)和Northstar-3(HGB-212)3期研究的受试者都已经接受ZYNTEGLO的治疗。HGB-212的后续工作正在进行中。 蓝鸟生物也在进行一项长期随访研究,LTF-303,以监测在输血依赖型β-地中海贫血症患者中ZYNTEGLO的安全性和有效性。
- 03.Hemgenix:美国FDA批准首款治疗血友病B成人患者的基因疗法!全球定价最贵
2022年11月22日,美国FDA宣布批准基因疗法Hemgenix(etranacogene dezaparvovec)上市,这是FDA批准的首款治疗血友病B成人患者的基因疗法!此疗法由荷兰生物技术公司UniQure开发,将由澳大利亚制药公司CSL Behring销售,将其定价为350万美元。

Hemgenix是一款基于AAV5载体的基因疗法,该药物搭载有凝血因子IX(FIX)基因变体(FIX-Padua),通过静脉给药,给药后该基因可在肝脏中表达FIX凝血因子,分泌后进入血液发挥凝血功能,从而达到治疗目的,理论上一次给药长期有效。
试验数据: 在两项针对Hemgenix的临床研究中,对57名18至75岁患有重度或中重度血友病B的成年男性进行了药物安全性和有效性评估,其有效性是根据男性年出血率(ABR)的下降情况确定的。在一项有54名参与者的研究中(HOPE-B trial),与基线相比,受试者的FIX活性增加,对常规FIX蛋白替代预防治疗的需求减少,ABR降低。此外,与Hemgenix相关的最常见不良反应包括肝酶升高、头痛和流感样症状等。
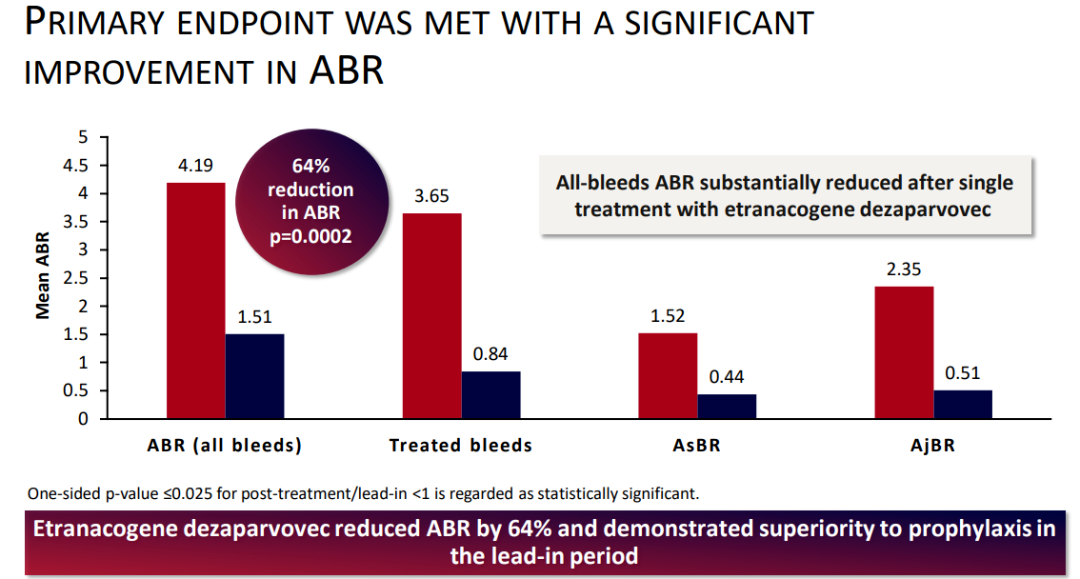
HOPE-B研究主要终点数据(来源:uniQure)
“血友病的基因治疗已经出现了20多年。尽管血友病的治疗取得了进展,但出血事件的预防和治疗会对个人的生活质量产生不利影响”美国FDA的CBER(生物制品评估和研究中心)主任彼得马克斯说。“今天的批准为血友病B患者提供了一种新的治疗选择。”
- 04.Adstiladrin:首款膀胱癌基因疗法
2022年12月16日宣布,美国FDA已批准Ferring Pharmaceuticals的基因疗法nadofaragene firadenovec-vncg上市,用于治疗伴有或不伴状瘤的高风险卡介苗(BCG)无反应的非肌肉浸润性膀胱癌(NMIBC)伴有或不伴状瘤的成年患者。

Adstiladrin(nadofaragene firadenovec-vncg)是一种基因疗法,用于治疗BCG无反应NMIBC的成年患者。它是一种基于非复制性腺病毒载体的基因疗法,含有基因干扰素α-2b,每三个月通过导管进入膀胱一次。载体进入膀胱壁的细胞,释放活性基因以完成其工作。细胞的内部基因/DNA机制“拾取”基因并翻译其DNA序列,导致细胞分泌大量干扰素α-2b蛋白,这是一种人体用来对抗癌症的天然蛋白质。因此,这种新颖的基因治疗方法将患者自身的膀胱壁细胞转化为干扰素微工厂,增强了人体对癌症的自然防御能力。
团队介绍: 美国FDA的批准是基于III期临床试验积极结果。 该研究共纳入157例患者,其中98例原位癌患者伴有/不伴有高级别Ta或T1(CIS±Ta/T1)。经过3个月1次Adstiladrin治疗后,有51%(50/98)患者实现了完全缓解(CR);在达到初始CR的患者中,有46%(23/50)在12个月时继续保持无高级别复发状态。研究中观察到的最常见的不良事件(AE)为:滴注部位分泌物(33%)、疲劳(24%)、膀胱痉挛(20%)、尿急(19%)和血尿(17%)等。因AE导致的停药率为1.9%。
二、首个成功出海的本土CAR-T细胞疗法:Carvykti
2022年2月28日,传奇生物(NASDAQ:LEGN)在美国新泽西州萨默塞特正式宣布,其自主研发的细胞治疗产品西达基奥仑赛(英文商品名:CARVYKTI®,英文通用名ciltacabtagene autoleucel,简称Cilta-cel)获得美国FDA批准上市,用于治疗复发或难治性多发性骨髓瘤(R/R MM)患者,这些患者既往接受过四种或四种以上的治疗,包括蛋白酶体抑制剂、免疫调节剂和抗CD38单克隆抗体。传奇生物与杨森公司于2017年12月签订了全球独家许可和合作协议,以开发和商业化CARVYKTI®。
Cilta-cel是一种靶向B细胞成熟抗原(BCMA)的嵌合抗原受体T细胞(CAR-T)疗法,使用嵌合抗原受体(CAR)的转基因对患者自身的T细胞进行修饰,以识别和消除表达BCMA的细胞。BCMA主要表达于恶性多发性骨髓瘤B细胞、晚期B细胞和浆细胞的表面。CARVYKTI®的CAR蛋白具有两种BCMA靶向单域抗体,对表达BCMA的细胞具有高亲和力,在与BCMA表达细胞结合后,CAR可促进T细胞活化、扩增,继而清除靶细胞。

试验数据: 在关键的CARTITUDE-1研究中,97例R/R MM患者出现了早期、深度持久的缓解,总缓解率(ORR)高达98%(95%CI:92.7-99.7),78%的患者获得了严格的完全缓解(sCR,95%CI:68.8-86.1)。在18个月的中位随访时间中,中位缓解持续时间(DOR)为21.8个月(95%CI,21.8-无法预估)。CARVYKTI®只能通过风险评估和缓解策略(REMS)的受限给药计划(CARVYKTI®REMS)获取。 CARVYKTI®的安全信息包括细胞因子释放综合征(CRS)、免疫效应细胞相关神经毒性综合征(ICANS)、帕金森病和吉兰-巴雷综合征、嗜血细胞性淋巴组织增生症/巨噬细胞活化综合征(HLM/MAS)以及长期或复发性细胞减少症的黑框警告。警告和预防措施包括长期复发性细胞减少、感染、低丙种球蛋白血症、超敏反应、继发性恶性肿瘤、对驾驶和使用机器的能力的影响。最常见的不良反应(≥20%)为发热、CRS、低丙种球蛋白血症、低血压、肌肉骨骼疼痛、疲劳、不明病原体感染、咳嗽、寒战、腹泻、恶心、脑病、食欲下降、上呼吸道感染、头痛、心动过速、头晕、呼吸困难、水肿、病毒感染、凝血功能障碍、便秘和呕吐。
三、两款疫苗:Spikevax新冠疫苗和Priorix麻腮风疫苗
- 01.Spikevax:美国FDA正式批准的第二款mRNA新冠疫苗
“Spikevax”是美国Moderna针对新型冠状病毒的mRNA疫苗,2020年12月18日,该疫苗获得美国FDA的紧急使用授权(EUA),用于预防18岁以上人群感染新冠病毒。2022年01月31日,FDA正式批准了该疫苗用于18岁以上的人群感染新冠病毒。

试验数据: 这一批准是基于美国FDA对正在进行的,支持该公司EUA的随机、设盲、含安慰剂对照临床试验的后续安全性和有效性数据的评估和分析。以及EUA之后的真实世界数据,以进一步检验疫苗的安全性和有效性。确定Spikevax有效性的最新分析包括14287例疫苗接种者和14164例安慰剂接种者,这些接种者为18岁以上,在接受首次接种前无新冠病毒感染证据。用于分析的数据是在Omicron变体出现之前积累的。 这些数据表明,Spikevax预防出现症状的COVID-19的有效率为93.2%,其中18-65岁人群有效率为93.4%,65岁以上人群有效率为91.5%。疫苗组发生55例COVID-19病例,安慰剂组发生744例COVID-19病例。该疫苗在预防严重疾病方面的有效率也达到了98.2%。主要副作用包括注射部位反应、关节痛、呕吐、疲劳、头痛、肌肉痛、寒颤、注射部位膨胀、发热等。
- 02.Priorix:麻腮风疫苗
2022年6月3日,美国FDA批准葛兰素史克麻疹、腮腺炎和风疹疫苗Priorix上市,用于在12个月以上的儿童中预防麻疹、腮腺炎和风疹(MMR),这是美国FDA批准的第二款MMR疫苗。首款MMR疫苗在1971年获批,来自默沙东。

Priorix的安全性在六项临床研究中进行了评估,共有12151名参与者(美国6391名)接受了至少一剂Priorix:8780名12至15个月大的儿童(美国4148名);2917名4至6岁儿童(美国1950名);454名7岁及以上的成年人和儿童(美国293名)。研究中出现的最常见不良反应是疼痛、发红、肿胀、食欲不振、易怒、嗜睡和发热。Priorix的有效性是通过与对照疫苗的免疫原性进行比较来证明的。
由于病例数量在近年不断增加,有相关机构预测麻疹疫苗市场到2026年可能达到64.1亿美元,市场潜力巨大。
四、首款经美国FDA批准的粪便微生物组疗法:Rebyota
2022年11月30日,美国FDA批准由Ferring Pharmaceuticals与旗下Rebiotix公司所开发的微生物组疗法Rebyota(fecal microbiota,live-jslm)用于降低18岁以上成年人艰难梭菌感染(CDI)后的复发。
这是首款经FDA批准的粪便微生物组疗法。在今年9月,美国FDA的疫苗与相关生物制品产品咨询委员会(VRBPAC)成员便以13:4的投票结果支持此疗法的批准。

一项关于粪菌移植治疗复杂性CDI有效性评价的随机临床研究数据显示,有22例患者接受经结肠镜异体粪菌移植,其中20例(90.9%)获得临床治愈,有24例接受自体粪菌移植仅有15例(62.5%)获得临床治愈(P=0.042),接受自体粪菌移植的患者中有9例出现了复发性CDI,上述9例患者在接受了异体粪菌移植后均无CDI的复发。由此可见,粪菌移植在治疗CDI中具有较高的有效性。本病例中,患者在经过甲硝唑及利福昔明治疗失败后,接受了粪菌移植治疗,且治疗后第2天即出现临床症状的缓解,也证实了粪菌移植在治疗难治性CDI中具有较好的疗效。
想要解锁更多药物研发信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、申报审批情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!

—END—











 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论