































确认和验证是一组相近的概念,确认(Qualifications)是证明厂房、设施和设备能够正确运行并且达到预期效果的一系列活动及其书面记录;验证(Validation)是证明任何操作规程或方法、生产工艺或系统能够达到预期效果的一系列活动及其书面记录。因此二者的内涵是基本一致的,只是确认侧重于硬件,验证侧重于软件。但需要注意的是,在验证活动中,也包含一种针对于软件的“确认(Verification)”,它强调的是某个规程或方法的符合性证明,与验证(Validation)在含义上有些微妙的区别。药品生产活动涉及到的主要确认与验证活动包括:厂房确认(Qualification of Facilities)、设施确认(Qualification of Utilities)、设备确认(Qualification of Equipment)、工艺验证(Process Validation)、包装验证(Validation of Packaging)、运输确认(Verification of Transportation)、分析方法验证(Validation of Test Methods)、分析方法确认(Verification of Test Methods)、清洁验证(Cleaning Validation)、清洁确认(Cleaning Verification)、计算机化系统验证(Validation of Computerized Systems)等。本节将选取连续制造中涉及到的较为典型的确认与验证活动进行分析论述。
1、设备确认
设备确认的目的是根据拟生产药品的工艺,对设备的设计和选型、安装与运行、对产品的适用性等做出评估,同时也为制定设备使用SOP、维护SOP奠定基础。设备确认的步骤包括“4Q”,即DQ(Design Qualification,设计确认)、IQ(Installation Qualification,安装确认)、OQ(Operational Qualification,运行确认)和PQ(Performance Qualification,性能确认),按顺序依次进行。其中对于一些原理简单、功能单一,或“即插即用”型的设备,常把IQ和OQ合并进行,称为IOQ。“4Q”确认的基本内容如表1所示。

对于连续制造,由于其批量具有很大程度的可变性,因此一条生产线甚至可以满足实验室规模、中试规模直至商业化规模所需的生产批量范围,设备的服务周期也可能从研发早期就已经开始。中国和国际主流GMP并不要求对实验室研发和中试批次的设备进行4Q确认,但考虑到设备生命周期的前移,设备确认活动也应当适当前移。对于有成熟质量管理体系的制药企业,建议其设备采购前就做好URS,设备入场安装时做好DQ和IQ,在设备运行前做好OQ,在工艺参数基本确定后开展PQ。对于质量体系尚未建立的初创企业和中小型企业,由于无法在研发早期开展完善的质量保证活动,因此应当在设备供应商比选时,选择有资质、有制药行业经验的设备供应商,在供应商协助下开展非受控体系下的4Q活动,同时应当保存好设备的FAT(Factory Acceptance Test,工厂验收测试)和SAT(Site Acceptance Test,现场验收测试)文件和所有的技术文件,以备后期追溯。一种连续制造和批生产的4Q时间节点设计方式如图1所示。与传统设备确认相比,连续制造的设备确认大幅前移,在小试阶段就可以完成3Q确认,在中试时,如果设备运转稳定,即可开展PQ。工艺放大至商业规模时,根据工艺变更情况,可以选择是否开展再确认活动。
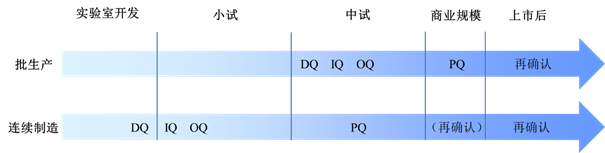
图1 批生产和连续制造的设备4Q时间节点设计
设备4Q的具体内容包括:在DQ阶段需要确认设备的设计是否能实现预期的连续制造功能,设别的工作原理是否能够持续稳定地输出符合要求的物料,设备的材质是否不污染物料和环境,是否易清洗,设备的结构和部件是否利于物料的转化和传递、利于清洁和维护,是否预留了必要的参数测试点和验证孔等。IQ阶段需要确认设备是否按照设计方案进行生产,配件、技术资料是否齐全,设备上的测量仪表是否能够符合精密度和准确度的要求,安装位置是否符合生产工艺,安装条件是否符合洁净要求,电源的额定电压和接地是否正确,支持设备运转的公用设施,比如制药用水、空压、氮气等是否正确连接,该设备和工艺上下游设备是否正确连接等。OQ阶段需要根据拟定规程对设备进行试验,以确认设备能否按照预期的工艺正常运行,确认在可能的最坏条件下设备能否正常开停机和运转,确认设备参数的可调节范围以及正常运行时的参数波动范围,确认设备仪表面板的操作可靠性,确认该设备与工艺上下游设备的物料转运速率是否协调等。PQ阶段需要在具体的工艺条件下,确认设备负载生产的运行稳定性、耐用性和重现性,确认所生产的产品质量的稳定性,确认在苛刻生产条件下(如工艺参数边缘)是否能够生产出符合要求的产品,确认该设备与上下游设备运转的协调性和流畅性等。由于连续制造的放大效应与批生产相比较不明显,因此在中试时就可以基本确定工艺参数,即可以开展PQ。如果放大到商业规模批量,关键工艺参数没有发生变更,则不需采取进一步的设备确认活动,否则应当开展再确认。在商业化生产期间,也应当定期开展再确认,以保证设备全生命周期的适用性。设备的再确认周期可由风险评估决定。
2、计算机化系统验证
计算机化系统是用于报告或者自动控制的集成系统,包括自动化的生产设备和检验设备、相关的信息和数据管理系统等。对计算机化系统开展验证活动的目的是确认计算机化系统符合用户需求和预期用途,保证由系统获取、处理、报告或存储的数据的可信性,降低数据完整性的风险。计算机化系统能够产生、储存、运算、输出大量的电子数据,而这些数据正是药品生产和检验活动的最原始记录。因此对于计算机化系统,首先应当保证系统时间的真实性、准确性与一致性;应当定期备份数据,备份及恢复流程必须经过验证,数据的备份及删除应有相应记录。在系统变更、升级或退役时,在整个系统的生命周期内需要保证原系统数据在规定的保存期限内能够进行查阅与追溯。
计算机化系统验证程度取决于待验证系统的复杂程度和预期用途。ISPE(International Society for PharmaceuticalEngineering,国际制药工程协会)于2008年制定了《良好自动化生产实践指南第五版》(Good Automated Manufacturing Practice 5,GAMP5),其对计算机化系统进行了分类,如表2所示。

如果一个计算机化系统是某个生产流程的组成部分(比如某些SCADA系统),一般不需要对该系统进行单独的验证,而是合并至该系统所依附的硬件设备确认中,因此本节尽管与上一节分列,这并不代表计算机化系统验证和设备确认一定是相互独立而无关联的。计算机化系统验证的流程与设备确认类似,也经历URS、DQ、IQ、OQ、PQ、再验证等阶段。在DQ中需要根据URS确认软件功能描述和版本、硬件与操作系统的要求、宏/公式/控制命令的规范、数据输入和输出的规范等;IQ中需要确认安装时的系统资源、可访问的用户(组)清单、与其他设备/接口的连接测试等;OQ中需要进行权限测试、报警测试、数据处理准确性测试等,可以用一组已知结果的输入数据进行测试,检验输出数据的准确性;PQ需要根据计算机化系统的功能进行区分,比如支持生产的系统可在连续3批生产过程中确认系统能否支持生产处合格的产品、支持检验的系统可以确认能否准确地使用某个分析方法得到正确的检验结果、其他系统可以根据其功能进行有目的的测试。
3、工艺验证
工艺验证是通过收集数据和评价数据,证实某一工艺过程能够始终如一地生产出符合预期要求和质量标准的产品的活动,采用某种工艺进行商业化生产前,必须验证其适用性,通过工艺验证确定工艺参数范围,支持制订和修订工艺规程。广义的工艺验证包括从工艺设计阶段一直到商业化生产阶段的整个过程,分为工艺设计(Process Design)、工艺性能确认(Process Performance Qualification,PPQ)和持续的工艺确认(Continued Process Verification,CPV (US),or Ongoing Process Verification (EU))三个主要阶段。无论哪一阶段,知识管理和质量风险管理的正确运用都是工艺验证成功的必要条件。
工艺设计是定义商业化生产工艺的阶段,也是起草工艺规程、批生产记录和批检验记录的阶段。工艺设计不需要在GMP条件下进行,但需要遵循科学的原则、设计方法和一定的文件编制规范。良好的工艺设计依赖于产品开发阶段获得的工艺知识和工艺理解,这些信息和数据可以通过DOE、小试或中试期间进行的实验来获得。尤其是对于某些单元操作和动力学的计算机模拟可以提供额外的工艺理解,以避免商业化放大可能出现的问题。对于工艺设计的评估即为PPQ阶段的主要内容。PPQ的执行需要结合实际的厂房、设施、设备、工艺、控制程序和经培训的人员,只有全部的要素都经过确认符合要求,才能确保PPQ结果的可靠。一个完整的PPQ方案应当至少包括:生产条件、数据的收集和评价方法、所需开展的检测和标准、分析方法的验证状态、取样方案、统计学方法的描述、先前未完成的硬件确认和人员培训(如有)等。PPQ必须按照商业化的生产工艺和程序实施,包括采用日常生产的人员、正常条件运转的公用设施和设备、符合GMP要求的文件系统等。PPQ的方法包括传统工艺验证方法(即前验证)、同步验证、持续性工艺确认(Continuous Process Verification)、混合方法等。各国法规对于PPQ执行的批次数量要求不尽相同,但业界通常的做法和共识是连续3个验证批的成功,即意味着PPQ的成功。PPQ的完成和报告的批准,标志着工艺进入商业生产阶段,工艺生产的产品也符合商业流通的质量要求。在产品获批后,工艺验证就进入第三阶段CPV,此阶段需要建立科学的程序和统计学方法以持续收集和评估与质量相关的工艺数据,探测出非期望的工艺变异,发现工艺的潜在问题并预防问题,使工艺保持已验证的状态。
与批生产相比,连续制造的工艺验证三阶段更具有同步性和普遍联系性,正如3.4.1节所述。连续制造的工艺验证和设备确认的联系更紧密,互动更强。这正是由于连续制造工艺在研发阶段即使用和商业化生产相同的设备,因此,设备确认也由批生产的PPQ阶段前移至连续制造的工艺设计阶段。对于连续制造的PPQ,由于其生产周期可能很长,因此应当更多关注工艺条件的稳定性和稳健性、产品的批内和批间一致性,关注工艺数据的实时收集和评估能力以及检测工艺偏移的能力。应当将生产线上的各个工艺设备作为一个整体来看待,而不是独立运行的个体,比如需要关注失重式喂料机的质量流速波动对整个工艺的影响等。连续制造PPQ的验证时间应当尽可能长,并且覆盖商业化生产的最大运行时间。
只有长时间运行才能发现许多有关工艺和设备的潜在问题,比如工艺漂移、设备疲劳、物料堆积等,并且能够获得产品批内一致性的证据。PPQ应当人为地设计一些干扰因素以研究真实生产可能发生的情况,比如更换在线检测所使用的探头、喂料机断料后加料、更换设备螺杆等。与批生产的PPQ批数要求相类似,连续制造也建议以至少连续3批的成功作为PPQ成功的标志。此外,Bryder等人和Ajay等人也提出了基于风险和统计学的批次数量确定方法。除了传统的前验证方法,持续性工艺确认作为PPQ的方法之一,也越来越应用于连续制造中。通过过程分析技术持续地监测工艺性能,并将工艺监测数据与工艺动态控制系统相结合,再通过调整流程来保证输出产品的质量。持续性工艺确认能更好地保持产品的批内一致性,且来自生产批次的大量数据支持成功验证的有力证据,因此持续性工艺确认是连续制造PPQ的理想方法之一。在CPV阶段需要首先基于风险,制定一套完整的方案,包括分析的执行频率、需要监测和分析的数据范围、合适的统计学方法以及数据偏离的标准。连续制造的CPV依赖于完善的工艺监测技术和过程分析技术。通过定量方法和统计学手段,对工艺参数、设备能力指数、IPC数据、输入物料和输出物料的质量属性等等工艺数据进行收集、分析、追踪,持续监测产品的批内和批间一致性,以证明工艺在整个商业化生产中始终处于受控状态。
实验室研究、工艺设计、PPQ、CPV,直到产品退市,构成了生产工艺的生命周期。好的工艺设计和开发方法能够预测工艺中的变异源,从而建立合适的警戒限和行动限、建立合适的手段探测、控制和消除变异。在产品生命周期中,不可避免地会发生数次的技术转移,只有按照科学地开展工艺开发和放大,理解了工艺的特性和影响模式,才能在技术转移时解决潜在发生的问题。
参考文献
[1] EDQM.Validation of Computerised Systems - Core document[EB/OL].(2018-08-01)[2020-04-02].https://www.edqm.eu/sites/default/files/guidelines-omcl-computerised_systems-core_document-march2018.pdf
[2] ISPE. GAMP 5Guide: Compliant GxP Computerized Systems[S].FL: ISPE, 2008.https://ispe.org/publications/guidance-documents/gamp-5
[3] FDA. ProcessValidation: General Principles and Practices[EB/OL]. (2011-01-01)[2020-04-02].https://www.fda.gov/media/71021/download
[4] EuropeanCommission. EU Guidelines for Good Manufacturing Practice for MedicinalProducts for Human and Veterinary Use: Annex 15: Qualification andValidation[EB/OL]. (2015-08-30)[2020-04-02].https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2015-10_annex15.pdf
[5] PMDA. PMDAViews on Applying Continuous Manufacturing to Pharmaceutical Products forIndustry[EB/OL]. (2018-03-30)[2020-04-02]. https://www.pmda.go.jp/files/000223712.pdf.
[6] Bryder M,Etling H, Fleming J, et al. Topic 1—stage 2 process validation: determining andjustifying the number of process qualification batches[J]. ISPE discussionpaper: PV stage, 2015, 2.
[7] PazhayattilA, Alsmeyer D, Chen S, et al. Stage 2 Process performance qualification (PPQ):a scientific approach to determine the number of PPQ batches[J]. AAPSPharmSciTech, 2016, 17(4): 829-833.
[8] Markarion J.Process validation for continuous manufacturing processes[EB/OL].(2014-11-02)[2020-04-02]. http://www.pharmtech.com/process-validation-continuous-manufacturing-processes




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论