































1.5.1 本周全球TOP10创新药研发进展
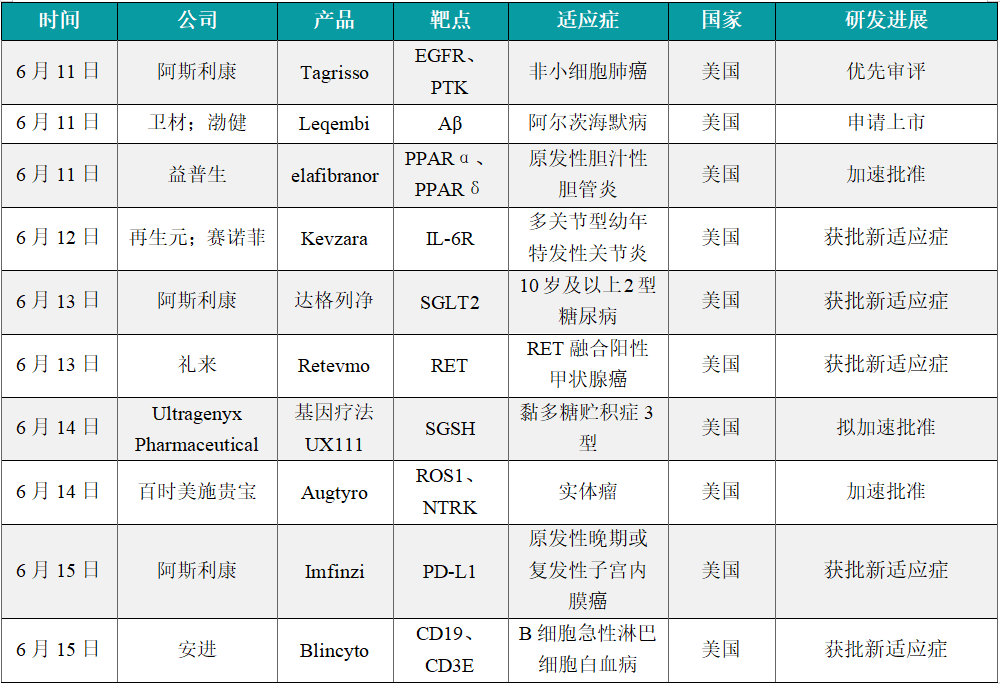
(1)阿斯利康小分子Tagrisso抑制剂获优先审评资格
6月11日,阿斯利康宣布其表皮生长因子受体(EGFR)酪氨酸激酶抑制剂(TKI)Tagrisso(osimertinib,奥希替尼)的补充新药申请(sNDA)已获美国FDA接受并授予优先审评资格,用以治疗接受放化疗(CRT)后无法切除的III期表皮生长因子受体突变(EGFRm)非小细胞肺癌(NSCLC)成人患者。如果获得批准,Tagrisso将适用于肿瘤具有外显子19缺失或外显子21(L858R)突变的EGFRm患者。FDA预计在2024年第四季度完成监管审评。FDA授予Tagrisso优先审评资格主要根据LAURA临床3期试验的结果。Tagrisso是第三代、不可逆的EGFR-TKI,已在临床证实对NSCLC具有疗效。
(2)卫材和渤健的Leqembi皮下注射剂型启动上市申请,治疗阿尔茨海默病
6月11日,卫材和渤健宣布,已经开始向美国FDA滚动提交阿尔茨海默病疗法Leqembi皮下注射剂型的生物制品许可(BLA)申请,每周一次作为维持疗法,治疗处于轻度认知障碍或轻度痴呆阶段的阿尔茨海默病患者。同时,美国FDA已接受每月一次静脉注射Leqembi的补充生物制品许可申请(sBLA),作为维持疗法治疗同一患者群体。FDA预计在明年初完成对该sBLA的审评。阿尔茨海默病是一种由有毒淀粉样蛋白引起的进展性疾病。这种病理过程将在患者的整个生命中持续进行,因此可能需要持续治疗,如果sBLA获得批准,通过每月一次的给药方案可以维持临床和生物标志物的益处,这种方案负担较小,更容易让患者和护理伙伴长期坚持。此外,Leqembi的皮下注射剂型使用自动注射器,也为患者和护理人员给药提供了方便。
(3)益普生的Iqirvo已获美国FDA加速批准,治疗成人原发性胆汁性胆管炎
6月11日,益普生宣布美国FDA已加速批准Iqirvo(elafibranor)80毫克片剂与熊去氧胆酸(UDCA)联合用于治疗对UDCA应答不足的成人原发性胆汁性胆管炎(PBC),或作为单药疗法治疗对UDCA不耐受的患者。Elafibranor是一种每日一次、口服、“first-in-class"双重过氧化物酶体激活受体(PPAR)α/δ激动剂,目前正在研究用于治疗罕见自身免疫性PBC患者。同时靶向活化PPAR α/δ可潜在治疗PBC的炎症、胆汁淤积和纤维化。2019年,美国FDA授予elafibranor突破性疗法认定,用于治疗对UDCA应答不佳的PBC成人患者。根据新闻稿,Iqirvo是近十年来首个获批用于治疗罕见肝病原发性胆汁性胆管炎的新药。益普生已向欧洲药品管理局(EMA)和英国药品和健康产品管理局(MHRA)提交监管申请,预计两者将在2024年下半年作出最终监管决定。
(4)再生元和赛诺菲共同开发的Kevzara获美国FDA批准,治疗多关节型幼年特发性关节炎
6月12日,再生元宣布,美国FDA批准其和赛诺菲共同开发的Kevzara(sarilumab),用于治疗体重63公斤或以上的活动性多关节型幼年特发性关节炎(pJIA)患者,这是一种同时影响多个关节的关节炎。美国FDA批准该疗法用于治疗pJIA患者群体是基于充分且具有良好对照的试验结果、来自类风湿性关节炎成年患者的药代动力学数据,以及针对pJIA儿童患者的药代动力学、药效学、剂量探索和安全性研究。Kevzara是再生元与赛诺菲共同开发的人源化单抗药物,它能结合白细胞介素-6(IL-6)受体,从而抑制由它介导的信号通路。除了pJIA,Kevzara目前已在25个国家或地区获批用于治疗中度至重度活动性类风湿性关节炎成人患者。Kevzara还获FDA批准用于治疗风湿性多肌痛。
(5)阿斯利康的达格列净已获得美国FDA批准,改善2型糖尿病儿童患者的血糖控制
6月13日,阿斯利康公司宣布Farxiga(达格列净)已获得美国FDA批准,用于改善10岁及以上2型糖尿病(T2D)儿童患者的血糖控制。此次FDA批准基于3期儿科临床试验T2NOW的积极结果,3期临床试验T2NOW的数据表明Farxiga可为T2D儿童和青少年患者提供具有临床意义的血糖改善。该患者群体中的安全性结果与成人T2D患者一致,符合Farxiga已知的安全性特征。Farxiga此前已在美国获批用于治疗成人T2D患者,作为饮食调节和运动的辅助措施来改善血糖控制。达格列净是一款“first-in-class”钠-葡萄糖协同转运蛋白2(SGLT2)抑制剂。截至2024年6月,它已在126个国家和地区获批,作为饮食调节和运动的辅助措施用于改善成人T2D患者的血糖控制。此外,它还在全球超过100个国家和地区获批用于治疗心力衰竭(包括射血分数降低型心力衰竭和射血分数保留型心力衰竭)和慢性肾病(CKD)成人患者。
(6)礼来的RET抑制剂Retevmo获完全批准,治疗RET融合阳性甲状腺癌
6月13日,美国FDA宣布,将礼来公司开发的RET抑制剂Retevmo(selpercatinib)的加速批准转化为完全批准,用于治疗晚期或转移性RET融合阳性甲状腺癌成人和2岁以上儿童患者。这些患者需要接受全身性治疗,并且对放射性碘疗法产生耐药性。Retevmo是一款强效RET激酶抑制剂,它在2020年首次获得美国FDA加速批准,治疗非小细胞肺癌(NSCLC)、MTC和甲状腺癌这三种癌症。这些患者肿瘤的RET基因出现融合或者突变。
(7)Ultragenyx Pharmaceutical的在研基因疗法UX111拟加速批准,治疗IIIA型黏多糖贮积症
6月14日,Ultragenyx Pharmaceutical宣布与美国FDA在会议中一致认为脑脊液(CSF)中的硫酸乙酰肝素(HS)是一个合理的替代终点,可用以支持其在研基因疗法UX111治疗IIIA型黏多糖贮积症(MPS IIIA)的生物制品许可申请(BLA)的加速批准。该公司将在与FDA的BLA前会议中最终确认BLA的细节,并计划在今年晚些时候或明年年初提交申请。MPS IIIA是一种常染色体隐性遗传病,由编码N-磺基葡萄糖胺磺基水解酶(SGSH)的基因发生突变所引起。UX111旨在使用AAV9载体,递送SGSH基因的功能性拷贝到中枢神经系统和外周器官,从而弥补SGSH酶的缺失。这次Ultragenyx与美国FDA达成的协议反映了FDA在加速罕见病症基因疗法审批方面的新动向。
(8)百时美施贵宝的Augtyro获美国FDA加速批准,治疗实体瘤
6月14日,百时美施贵宝宣布,美国FDA加速批准其口服酪氨酸激酶抑制剂(TKI)Augtyro(repotrectinib)用于治疗局部晚期、转移性或手术切除可能导致严重疾病、治疗后病情进展或没有令人满意的替代疗法的实体瘤成人和12岁及以上的儿童患者,这些患者的肿瘤为神经营养酪氨酸受体激酶(NTRK)基因融合阳性。Augtyro是Turning Point公司(现已被百时美施贵宝收购)开发的一款ROS1和NTRK靶向抑制剂。它具有独特的结构,与靶点蛋白的结合位点位于“ATP口袋”内,并且不受多种耐药性突变的影响。因此,它能够克服多种对其它TKI产生抗性的基因突变,杀死携带ROS1或NTRK基因融合的多种肿瘤细胞。该疗法于2023年11月获FDA批准用于局部晚期或转移性ROS1阳性非小细胞肺癌(NSCLC)成人患者的治疗。根据新闻稿,Augtyro是首款获批用于ROS1阳性NSCLC患者的新一代TKI疗法。
(9)阿斯利康的PD-L1抑制剂Imfinzi与卡铂和紫杉醇联用获FDA批准,治疗子宫内膜癌
6月15日,美国FDA宣布,批准阿斯利康公司的重磅PD-L1抑制剂Imfinzi与卡铂和紫杉醇联用,然后作为单药,用于治疗具有错配修复缺陷(dMMR)的成人原发性晚期或复发性子宫内膜癌患者。Imfinzi是一种人源化单克隆抗体,它与PD-L1蛋白结合并阻断PD-L1与PD-1及CD80蛋白的相互作用,从而对抗肿瘤的免疫逃逸策略,并解除对免疫反应的抑制,增强免疫系统杀伤癌细胞的能力。这一治疗方案的疗效得到DUO-E临床试验的支持,试验结果显示,虽然在总体人群中,Imfinzi联合卡铂和紫杉醇与单用卡铂和紫杉醇相比,在无进展生存期上有统计学显著的改善,但总体人群的改善主要归因于携带dMMR肿瘤的患者。
(10)安进的双特异性抗体疗法Blincyto获FDA批准,治疗B细胞前体急性淋巴细胞白血病
6月15日,安进宣布美国FDA批准其双特异性抗体疗法Blincyto(blinatumomab)用于治疗年龄一个月或以上、CD19阳性、费城染色体阴性B细胞前体急性淋巴细胞白血病(B-ALL)患者的巩固阶段治疗,无论患者的可测量残留病灶(measurable residual disease,MRD)状态如何。此次批准是Blincyto所获批的第三项适应症。该批准主要基于E1910临床3期试验的结果。该试验研究了新确诊的费城染色体阴性B-ALL患者接受诱导后巩固治疗的效果,旨在深化缓解以实现持久的应答。Blincyto是安进公司开发的双特异性T细胞接合器(BiTE),它的一端与B细胞表面表达的CD19抗原相结合,另一端与T细胞表面的CD3受体相结合。它能够将T细胞募集到癌细胞附近,促进它们对癌细胞的杀伤。Blincyto已经获得FDA批准治疗复发/难治性B细胞ALL患者。
1.5.2本周全球TOP10积极/失败临床结果
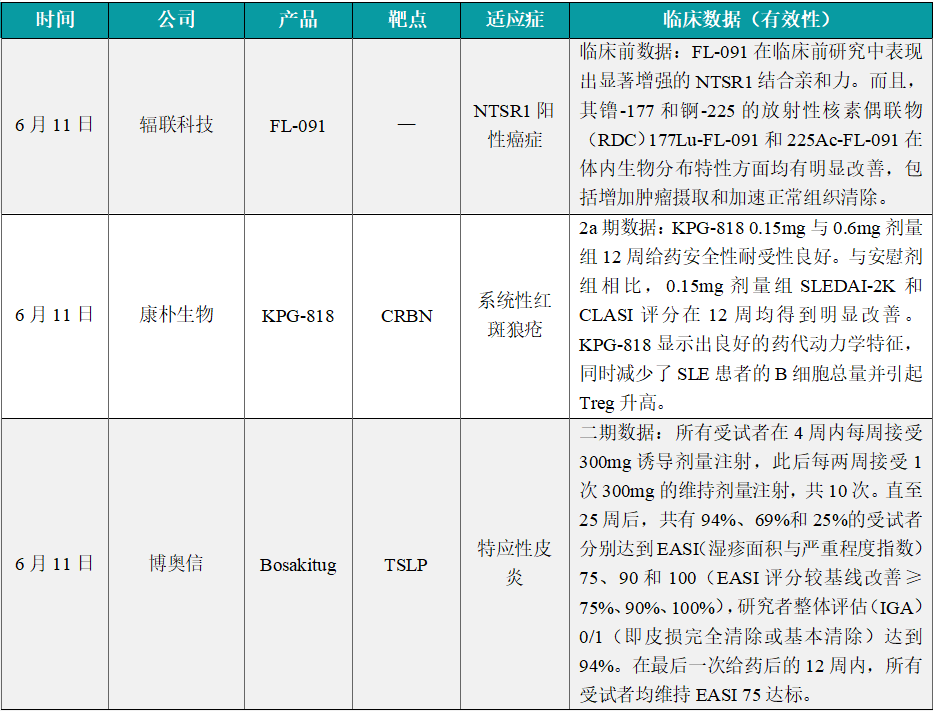
(1)辐联科技FL-091临床前研究显示NTSR1靶向放射性药物在癌症治疗中的潜力
6月11日,辐联科技在于加拿大举办的2024年SNMMI(核医学和分子影像学会)年会上以口头报告形式公布了其NTSR1靶向放射性核素偶联药物(RDC)项目FL-091的临床前数据研究成果。神经降压素受体1(NTSR1)蛋白在各种恶性肿瘤的发展、转移和预后中扮演着重要角色,被视为癌症治疗的前沿靶点之一。公开信息显示,FL-091在临床前研究中表现出显著增强的NTSR1结合亲和力。而且,其镥-177和锕-225的放射性核素偶联物(RDC)177Lu-FL-091和225Ac-FL-091在体内生物分布特性方面均有明显改善,包括增加肿瘤摄取和加速正常组织清除。更重要的是,这种生物分布特性的改善使其在不同的肿瘤异种移植模型中展现出优异的抗肿瘤活性。这些数据强有力地支持FL-091作为靶向NTSR1阳性癌症的有效放射性配体载体。目前,辐联科技正在进行对靶向NTSR1阳性癌症的α疗法候选药物225Ac-FL-091的开发。
(2)康朴生物KPG-818 2a期临床结果积极,治疗SLE
6月11日,康朴生物发布KPG-818用于治疗系统性红斑狼疮(SLE)的2a期临床研究结果摘要。SLE是机体免疫系统攻击自身组织导致的一种复杂的慢性自身免疫性疾病。根据康朴生物新闻稿,KPG-818是康朴生物自主开发的分子胶口服小分子免疫调节药物,归属E3泛素连接酶复合物CRL4CRBN调节剂,对靶点CRBN显示出极高的亲和力,可以高效降解锌指转录因子Aiolos(IKZF3)和Ikaros(IKZF1),有效调节免疫细胞(B细胞、T细胞和Treg细胞)以及相关免疫细胞因子的表达水平。研究结果显示,KPG-818 0.15mg与0.6mg剂量组12周给药安全性耐受性良好。与安慰剂组相比,0.15mg剂量组SLEDAI-2K和CLASI评分在12周均得到明显改善。KPG-818显示出良好的药代动力学特征,同时减少了SLE患者的B细胞总量并引起Treg升高。该项临床试验的研究者、佛罗里达风湿病学会前任主席Robert Levin医生表示,在本次研究中,KPG-818展现的初步疗效让人充满期待,在有皮肤症状的SLE患者中表现尤其亮眼。
(3)博奥信bosakitug 2期临床试验结果积极,治疗特应性皮炎
6月11日,博奥信宣布ADAMANT研究的关键结果。这是一项bosakitug(一种高效抗TSLP单克隆抗体)治疗特应性皮炎患者的临床2期概念验证(POC)试验。Bosakitug(BSI-045B/TQC2731)是一种高亲和力的人源化单克隆抗体,靶向胸腺基质淋巴细胞生成素(TSLP)。在会议上公布的试验显示,所有受试者在4周内每周接受300mg诱导剂量注射,此后每两周接受1次300mg的维持剂量注射,共10次。直至25周后,共有94%、69%和25%的受试者分别达到EASI(湿疹面积与严重程度指数)75、90和100(EASI评分较基线改善≥75%、90%、100%),研究者整体评估(IGA)0/1(即皮损完全清除或基本清除)达到94%。在最后一次给药后的12周内,所有受试者均维持EASI 75达标。
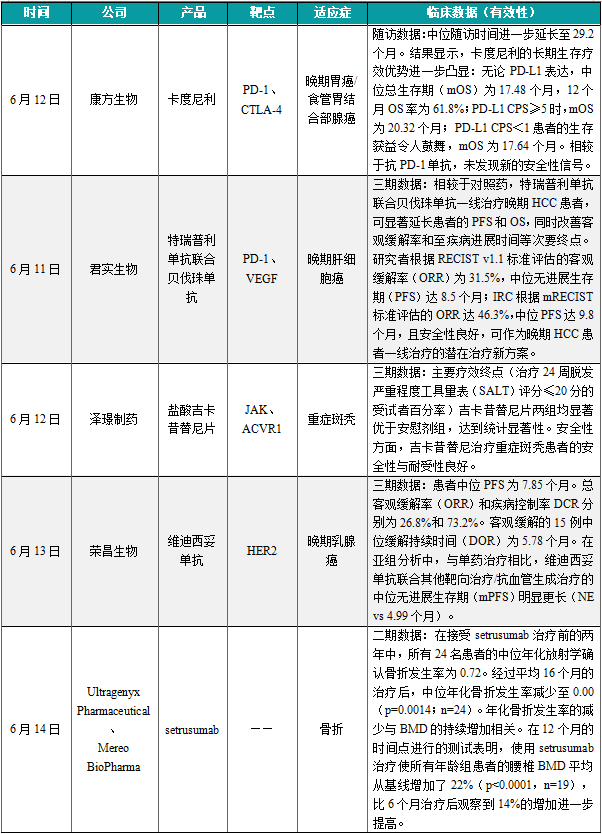
(4)康方生物PD-1/CTLA-4双特异性抗体1b/2期临床积极,治疗晚期胃癌
6月12日,康方生物宣布,其自主研发的PD-1/CTLA-4双特异性抗体卡度尼利单抗注射液联合化疗一线治疗不可手术切除的局部晚期、复发或转移性胃或胃食管结合部(G/GEJ)腺癌的1b/2期临床研究结果,本次发布数据的中位随访时间进一步延长至29.2个月,结果显示卡度尼利的长期生存疗效优势进一步凸显:无论PD-L1表达,中位总生存期(mOS)为17.48个月,12个月OS率为61.8%;PD-L1 CPS≥5时,mOS为20.32个月;PD-L1 CPS<1患者的生存获益令人鼓舞,mOS为17.64个月。相较于抗PD-1单抗,未发现新的安全性信号。康方生物新闻稿表示,扎实的临床循证医学研究表明,卡度尼利联合化疗方案可显著提高晚期胃癌全人群患者的总生存获益和降低疾病死亡风险。无论PD-L1表达,包括PD-L1高表达、低表达以及阴性患者,卡度尼利方案都展现了优异的疗效,有望弥补当下抗PD-1单抗在PD-L1低表达以及阴性胃癌中疗效有限的临床短板。
(5)君实生物特瑞普利单抗联合贝伐珠单抗3期结果积极,治疗晚期肝细胞癌
6月11日,君实生物宣布,其自主研发的抗PD-1单抗特瑞普利单抗联合贝伐珠单抗一线治疗晚期肝细胞癌(HCC)的3期HEPATORCH研究的主要研究终点无进展生存期(PFS,基于独立影像评估)和总生存期(OS)均已达到方案预设的优效边界。君实生物计划将于近期向监管部门递交该新适应症的上市申请肝癌是全世界范围内常见的消化系统恶性肿瘤,病理类型以HCC为主(约占90%)。特瑞普利单抗联合贝伐珠单抗一线治疗不可切除或转移性HCC的有效性和安全性。根据研究分析结果,相较于对照药,特瑞普利单抗联合贝伐珠单抗一线治疗晚期HCC患者,可显著延长患者的PFS和OS,同时改善客观缓解率和至疾病进展时间等次要终点。特瑞普利单抗联合贝伐珠单抗一线治疗晚期HCC显示出令人鼓舞的疗效和生存获益,研究者根据RECIST v1.1标准评估的客观缓解率(ORR)为31.5%,中位无进展生存期(PFS)达8.5个月;IRC根据mRECIST标准评估的ORR达46.3%,中位PFS达9.8个月,且安全性良好,可作为晚期HCC患者一线治疗的潜在治疗新方案。
(6)泽璟制药盐酸吉卡昔替尼片治疗重症斑秃3期临床试验达到主要疗效终点
6月12日,泽璟制药宣布其自主研发的1类新药盐酸吉卡昔替尼片(曾用名:盐酸杰克替尼片)治疗重症斑秃的3期临床主试验达到了主要疗效终点,达到统计显著性。泽璟制药表示将加快推进该产品重症斑秃适应症的上市进程。盐酸吉卡昔替尼是一种新型JAK和ACVR1双抑制剂类药物。本次达主要终点的研究在中国44家医院开展,符合方案要求的425例重症斑秃患者随机入组,分配到吉卡昔替尼片50mg Bid组、75mg Bid组或安慰剂片组,经独立第三方非盲团队对该项试验中完成24周治疗的数据进行分析后,结果显示主要疗效终点(治疗24周脱发严重程度工具量表(SALT)评分≤20分的受试者百分率)吉卡昔替尼片两组均显著优于安慰剂组,达到统计显著性。安全性方面,吉卡昔替尼治疗重症斑秃患者的安全性与耐受性良好。有关该项临床试验的详细数据,将在后续相关学术会议上公布。目前,吉卡昔替尼治疗重症斑秃3期临床研究的延伸试验正在进行中。
(7)荣昌生物维迪西妥单抗3期临床试验达到主要终点,计划递交上市申请
6月13日,荣昌生物宣布,靶向HER2的抗体偶联药物(ADC)维迪西妥单抗治疗HER2阳性存在肝转移的晚期乳腺癌患者的3期临床取得阳性结果,达到主要研究终点。荣昌生物计划近期向CDE递交上市申请。针对该适应症,维迪西妥单抗此前已于2021年6月被CDE纳入突破性治疗品种。维迪西妥单抗是荣昌生物研发的靶向HER2的抗体偶联药物(ADC),目前已有胃癌、尿路上皮癌两大适应症获批上市。今年5月,该产品联合特瑞普利单抗治疗肌层浸润性膀胱癌还被CDE纳入突破性治疗品种。研究结果显示,患者中位PFS为7.85个月。总客观缓解率(ORR)和疾病控制率DCR分别为26.8%和73.2%。客观缓解的15例中位缓解持续时间(DOR)为5.78个月。在亚组分析中,与单药治疗相比,维迪西妥单抗联合其他靶向治疗/抗血管生成治疗的中位无进展生存期(mPFS)明显更长(NE vs 4.99个月)。研究认为,维迪西妥单抗在HER2阳性晚期乳腺癌患者群体中显示出有希望的抗肿瘤活性和可接受的耐受性。
(8)Ultragenyx的setrusumab 2/3期临床结果积极,减少成骨不全患者骨折发生率
6月14日,Ultragenyx Pharmaceutical和Mereo BioPharma公司宣布,在研疗法setrusumab在治疗成骨不全(OI)患者的2/3期临床试验Orbit的2期部分中,在至少14个月的随访中继续显著减少患者的骨折发生率,中位年化骨折发生率为0。同时,使用setrusumab治疗还在第12个月显著且持续地提高了腰椎的骨密度(BMD)。数据显示,在接受setrusumab治疗前的两年中,所有24名患者的中位年化放射学确认骨折发生率为0.72。经过平均16个月的治疗后,中位年化骨折发生率减少至0.00(p=0.0014;n=24)。年化骨折发生率不包括形态学椎体骨折以及手指、脚趾、头骨和面部的骨折,这与3期研究的主要疗效终点一致。年化骨折发生率的减少与BMD的持续增加相关。在12个月的时间点进行的测试表明,使用setrusumab治疗使所有年龄组患者的腰椎BMD平均从基线增加了22%(p<0.0001,n=19),比6个月治疗后观察到14%的增加进一步提高。
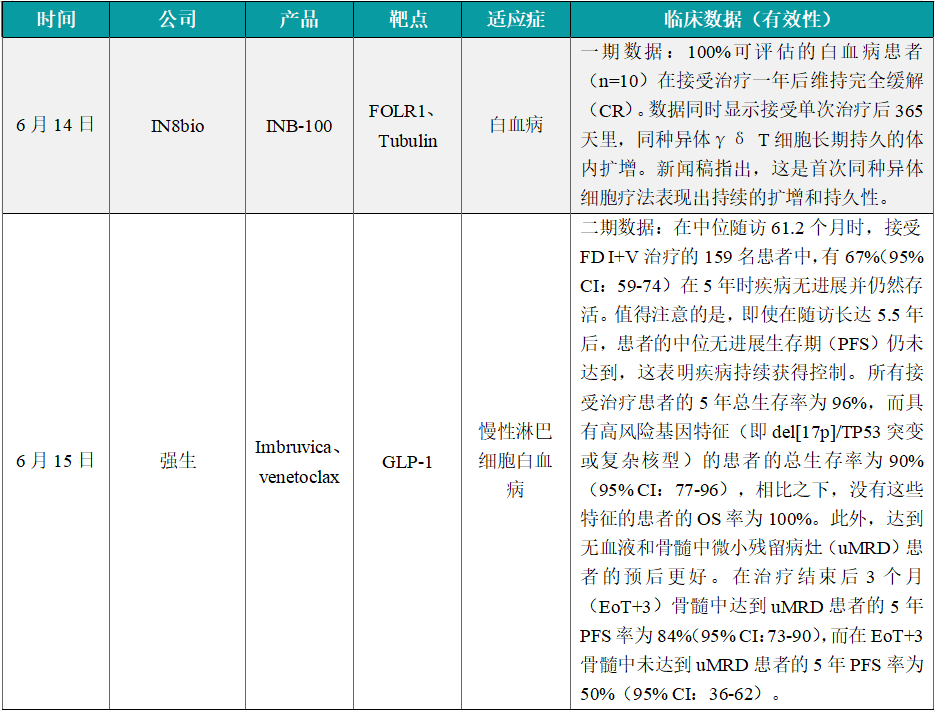
(9)IN8bio细胞疗法1期结果积极,治疗白血病
6月14日,IN8bio展示了其在研现货型γδ T细胞疗法INB-100的最新1期临床试验结果。试验结果显示,100%可评估的白血病患者(n=10)在接受治疗一年后维持完全缓解(CR)。数据同时显示接受单次治疗后365天里,同种异体γδ T细胞长期持久的体内扩增。新闻稿指出,这是首次同种异体细胞疗法表现出持续的扩增和持久性。INB-100是一款来自健康供体的同种异体γδ T细胞,在体外经过扩增和激活后,被输注到白血病患者体内。IN8bio计划在今年夏天与FDA会晤,讨论注册性临床试验的规划。
(10)强生的Imbruvica联合疗法2期结果积极,初治CLL患者5年总生存率高达96%
6月15日,强生宣布了CAPTIVATE临床2期研究的最新结果,该研究评估了固定疗程的Imbruvica(ibrutinib,伊布替尼)与venetoclax联合(I+V)用于初治慢性淋巴细胞白血病(CLL)患者的疗效。Imbruvica是一款口服、一日一次的布鲁顿氏酪氨酸激酶(BTK)抑制剂。结果显示,所有接受治疗患者的5年总生存率高达96%,在中位随访61.2个月时,接受I+V治疗的159名患者中,有67%(95% CI:59-74)在5年时疾病无进展并仍然存活。值得注意的是,即使在随访长达5.5年后,患者的中位无进展生存期(PFS)仍未达到,这表明疾病持续获得控制。所有接受治疗患者的5年总生存率为96%,而具有高风险基因特征(即del[17p]/TP53突变或复杂核型)的患者的总生存率为90%(95% CI:77-96),相比之下,没有这些特征的患者的OS率为100%。此外,达到无血液和骨髓中微小残留病灶(uMRD)患者的预后更好。在治疗结束后3个月(EoT+3)骨髓中达到uMRD患者的5年PFS率为84%(95% CI:73-90),而在EoT+3骨髓中未达到uMRD患者的5年PFS率为50%(95% CI:36-62)。
同期事件:
1. 2024年第24周06.10-06.16国内创新药/改良型新药申请临床/获批临床/申请上市/获批上市数据分析
2. 2024年第24周06.10-06.16国内仿制药/生物类似物申报/审批数据分析
3. 2024年第24周06.10-06.16国内医药大健康行业政策法规汇总
4. 2024年第24周06.10-06.16全球医药大健康行业投融资数据
以上内容均来自{摩熵咨询医药行业观察周报(2024.06.10-2024.06.16)},如需查看或下载报告,可点击!
<END>
想要解锁更多药物研发信息吗?查询摩熵医药数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!

















 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论