































2024年6月22日,和黄医药宣布,公司自主研发的抗肿瘤新药呋喹替尼获得欧盟委员会批准作为单药疗法用于治疗经治成人转移性结直肠癌(mCRC)患者。此前,2023年9月呋喹替尼已经成功在美国获批上市。截止目前,共有8款国产1类创新药在欧美日发达国家市场获批。
一、首款在美获批的国产新药:百济神州BTK抑制剂泽布替尼
2019年11月14日,泽布替尼在美国取得全球首次批准,实现中国抗癌新药出海“零的突破”,随后其陆续在中国、加拿大、澳大利亚、俄罗斯、欧盟等多个国家和地区获批上市,目前其商业化足迹已遍布全球40多个市场。此外,其在全球范围内仍有多项药政申报正在审评中。

2021年6月11日举行的26届欧洲血液学协会年会(EHA 2021)网络大会的主席研讨会上,百济神州发布的一项全球头对头3期临床研究最新数据表明:该公司自主研发的抗癌新药泽布替尼(商品名“百悦泽”),以更好的疗效和安全性,在数据上优胜强生公司研发的抗癌药伊布替尼。这是中国创新药首次在头对头中打败欧美跨国公司药物。2022年12月14日,百济神州公布,公司自主研发的BTK抑制剂泽布替尼(商品名:百悦泽®)在全球3期头对头研究ALPINE试验中无进展生存期(PFS)的终期分析结果。
经独立评审委员会(IRC)和研究者评估,泽布替尼对比伊布替尼,用于治疗复发/难治性(R/R)慢性淋巴细胞白血病(CLL)/小淋巴细胞淋巴瘤(SLL)患者,取得PFS的优效性(HR:0.65[95% CI,0.49 ~ 0.86],p = 0.0024)。24个月时,IRC评估的泽布替尼PFS率为79.5%,而伊布替尼则为67.3%。基于ALPINE研究,泽布替尼成为目前全球首个且唯一一款对比伊布替尼,取得PFS与总缓解率(ORR)双重优效性的BTK抑制剂。

百济神州2023年报显示,公司全年业绩再创历史新高,总收入达25亿美元,同比增长74%。全球产品收入持续攀升,创收22亿美元,同比增长75%。核心自研药物BTK 抑制剂泽布替尼(百悦泽)全球销售额首次突破十亿美元大关,达 13 亿美元,成为国内首个“重磅炸弹”产品。除此之外,PD-1 产品百泽安(替雷利珠单抗)逐渐放量,逐步攻克海外市场。
二、传奇生物:BCMA CAR-T产品西达基奥仑赛
西达基奥仑赛(Cilta-cel,CARVYKTI)是传奇生物开发的一种CAR-T疗法,在美国和欧洲之前被称为JNJ4528,而在中国则被称为LCAR-B38M细胞疗法。2017年12月传奇生物与强生子公司杨森达成了全球化合作和许可协议,共同对西达基奥仑赛进行开发、生产和商业化。传奇生物向强生授予在全球范围内共同开发和商业化西达基奥仑赛的许可,由传奇生物负责大中华区的价格批准和预订销售,由强生负责世界各地的价格批准和预订销售。根据协议,传奇生物将从杨森获得3.5亿美元的预付款,并有权在开发、生产、监管和销售方面达到里程碑进展时获得额外付款。该协议规定,在除大中华区以外的全球市场中,传奇生物和杨森公司的成本和利润分摊比例为50%/50%,在大中华区该分摊比例为70%/30%(传奇/杨森)。自此,西达基奥仑赛开启全球性研发、临床、生产的商业化征程。

西达基奥仑赛于2022年2月28日获得FDA批准上市,用于治疗复发或难治性多发性骨髓瘤成人患者,这是首款中国自主原创并在美国上市的CAR-T细胞治疗产品。此次获批是主要基于关键性临床Ib/II期CARTITUDE-1 (NCT03548207) 研究结果。最新数据显示,西达基奥仑赛在既往接受过四线或者以上治疗(包括蛋白酶体抑制剂、免疫调节剂和抗CD38单克隆抗体)的复发或难治性多发性骨髓瘤患者中显示出高达98%的总缓解率。在关键的CARTITUDE-1 (NCT03548207) 研究中,97例R/R MM患者出现了早期、深度持久的缓解,总缓解率(ORR)高达98%(95%CI:92.7-99.7),78%的患者获得了严格的完全缓解(sCR,95%CI:68.8-86.1)。在18个月的中位随访时间中,中位缓解持续时间(DOR)为21.8个月(95% CI,21.8-无法预估)。

2024年3月11日,金斯瑞旗下传奇生物发布2023年财务业绩。财报显示,传奇生物2023总营收为2.85亿美元,较上年同期的1.17亿美元同比增长144%。CARVYKTI®(西达基奥仑赛)的成功商业化助推了其业绩增长,作为传奇生物唯一一款商业化产品,CARVYKTI®2023年全球销售额达到5亿美元!
三、天济医药:全新机理外敷银屑病药物本维莫德
2022年5月24日,Dermavant Sciences宣布FDA已批准VTAMA(本维莫德,1%)乳膏上市,用于成人斑块型银屑病的局部治疗。本维莫德由国内企业天济医药研发、是具有自主知识产权的“first-in-class”首创新药。2012年,天济医药将本维莫德境外开发权授予GSK,合同首付款约2亿美元;2018年7月,GSK又以3.3亿美元的价格将本维莫德在中国境外的开发权出售给Dermavant公司。早在2019年5月,本维莫德以“1类新药”在国内获批上市,用于适合局部治疗的成人轻至中度稳定性寻常型银屑病。这也是目前首款我国先批准上市之后才获美国FDA批准的创新药。
在两项关键性III期临床试验(PSOARING 1和PSOARING 2)中,与对照组相比,第12周时VTAMA乳膏在医师总体评估(PGA)评分“皮肤症状完全清除”(PGA=0)或“几乎完全清除”(PGA=1)方面表现出高度统计学显著改善,并且评分至少提高两级的患者比例显著高于对照组,达到试验的主要终点。同时,VTAMA乳膏也达到了所有关键次要终点,包括PASI75(银屑病面积与严重性指数评分改善75%)。
四、君实生物:首款在美获批的国产PD-1产品特瑞普利单抗
2023年10月28日,Coherus BioSciences与君实生物宣布,美国FDA批准其单抗Loqtorzi(toripalimab,特瑞普利单抗)联合吉西他滨/顺铂作为晚期复发或转移性鼻咽癌(NPC)患者的一线治疗。其单药也获批用于复发或转移性鼻咽癌含铂治疗后的二线及以上治疗。特瑞普利单抗是首个FDA批准用以治疗鼻咽癌的PD-1单抗。

特瑞普利单抗(中文商品名:拓益)是一款以PD-1为靶点的单抗药物,其上市申请是基于JUPITER-02(一项随机、双盲、安慰剂对照、国际多中心3期临床研究)及POLARIS-02(一项多中心、开放标签、2期关键注册临床研究)的研究结果。
在JUPITER-02临床3期研究中,与单独化疗相比,特瑞普利单抗联合化疗显著改善患者的无进展生存期(PFS),将疾病进展或死亡风险降低了48%。该药物还显示总生存期(OS)出现具有统计学显著性和临床意义的改善,与单独化疗相比,特瑞普利单抗导致死亡风险降低37%。JUPITER-02研究结果于2021年6月在美国临床肿瘤学会(ASCO)年会的全体大会(#LBA2)上首次发表,随后作为《自然-医学》(Nature Medicine)2021年9月刊的封面文章发表。
POLARIS-02研究共纳入190例复发或转移性鼻咽癌患者接受特瑞普利单抗单药治疗,结果显示,在ITT人群中(N=190),客观缓解率(ORR)为20.5%,中位缓解持续时间(DOR)为12.8个月,中位总生存期(OS)为17.4个月;在92例既往接受过至少2线系统化疗失败的患者中,ORR为23.9%,中位缓解持续时间(mDOR)达到14.9个月,疾病控制率(DCR)为41.3%,中位总生存时间(mOS)达到15.1个月。2021年1月POLARIS-02研究在国际著名期刊《临床肿瘤学杂志》(Journal of Clinical Oncology,IF: 32.956)发表。

君实生物合作伙伴Coherus对特瑞普利单抗的销售表示乐观,认为鼻咽癌适应症在两年后能够达到销售额峰值2亿美元。抗PD-1单抗药物特瑞普利单抗治疗鼻咽癌获得了FDA的孤儿药(用于预防、治疗、诊断罕见病的药品)资格和突破性疗法认定,美国2024年鼻咽癌新发病例约为2000例。
五、和黄医药:口服VEGFR1/2/3抑制剂呋喹替尼
2023年11月9日(美国时间11月8日)和黄医药宣布,其与合作伙伴武田共同开发的药物Fruzaqla(呋喹替尼/fruquintinib)在美国获批进入当地医药市场,用于治疗晚期结直肠癌。这是美国首个且唯一获批用于治疗经治转移性结直肠癌的,针对全部三种抗血管内皮生长因子(VEGF)受体的高选择性抑制剂。

FRUZAQLA(呋喹替尼)是一种选择性的口服VEGFR-1、-2及-3抑制剂。VEGFR抑制剂在抑制肿瘤的血管生成中起到至关重要的作用。FRUZAQLA被设计为拥有更高的激酶选择性,旨在降低脱靶激酶活性,从而实现更高的药物暴露、对靶点的持续覆盖以及当潜在作为联合疗法时拥有更高的灵活度。迄今为止,FRUZAQLA展示出可控的安全性特征,其与其他抗肿瘤疗法联合使用的研究正在进行中。
2007年,呋喹替尼在位于上海浦东张江的实验室首次合成最初的一个小分子结构,2018年在中国获批上市。今天,终于实现了在美国的成功上市!该药物于2020年1月纳入国家医保目录,目前市场已覆盖全国328个城市,超过3000家肿瘤医院。根据药融云全国医院销售数据,截至2023年,该药物在院内累计销售近13亿元人民币。资料显示,呋喹替尼在三线结直肠癌患者中的市场占有率达47%,惠及逾8万名结直肠癌患者。
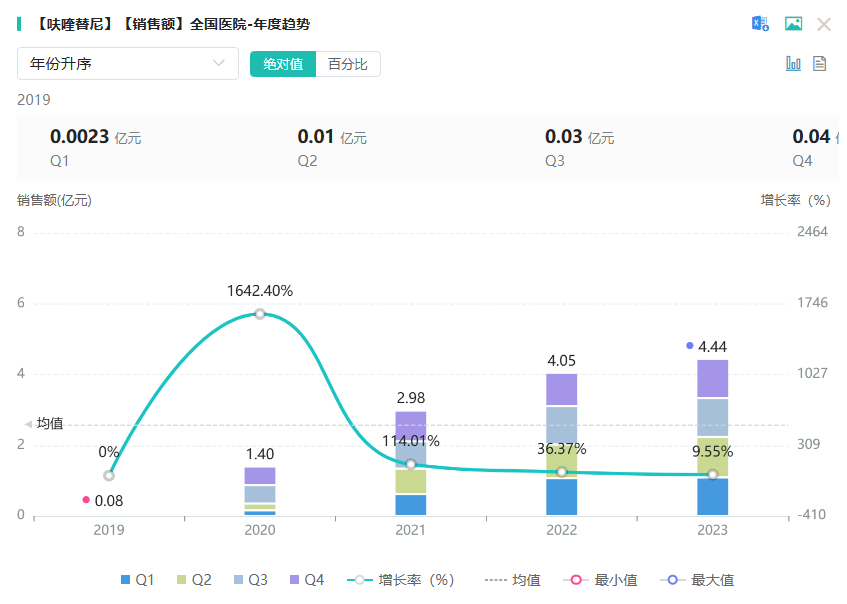
来源:药融云全国医院销售数据
FRUZAQLA的获批是基于两项大型III期临床试验的数据,包括:国际多中心临床试验FRESCO-2研究,其数据亦已于《柳叶刀 (The Lancet) 》上发表;以及于中国开展的FRESCO研究,其数据亦已于《美国医学会杂志 (JAMA) 》上发表。上述研究探索了FRUZAQLA联合最佳支持治疗对比安慰剂联合最佳支持治疗用于治疗经治转移性结直肠癌患者。FRESCO及FRESCO‑2研究均达到了其主要终点及关键次要终点,并在总共734名接受FRUZAQLA治疗的患者中展现出了一致的获益。各项研究的安全性特征亦保持一致。
FRESCO及FRESCO-2研究的数据亦支持了向欧洲药品管理局("EMA")提交的呋喹替尼上市许可申请,该申请已于2023年6月获确认及受理。此外,一项向日本医药品和医疗器械局("PMDA")的申请亦于2023年9月提交。2024年6 月 22 日,和黄医药宣布,公司自主研发的抗肿瘤新药呋喹替尼获得欧盟委员会批准作为单药疗法用于治疗经治成人转移性结直肠癌(mCRC)患者。
六、百济神州:第二款在美获批的国产PD-1产品替雷利珠单抗
2023年9月15日,替雷利珠单抗获得欧洲药品管理局(EMA)人用药品委员会(CHMP)批准并在欧盟上市,用于治疗食管癌;2024年2月获批治疗3种适应证的非小细胞肺癌,优良疗效获国际认可。2024年3月14日,替雷利珠单抗(百泽安,TEVIMBRA)获得美国FDA的批准,用于治疗既往经系统治疗后不可切除的、复发性局部晚期或转移性食管鳞状细胞癌(ESCC)患者。
替雷利珠单抗是一款人源化免疫球蛋白G4单克隆抗体,对PD-1具有高亲和力和结合特异性,其可以减少与巨噬细胞上Fcγ受体的结合,从而帮助免疫细胞识别并对抗肿瘤。
在欧、美获批主要基于III期RATIONALE 302研究(NCT03430843)的积极结果,其是一项全球、随机、开放性试验,旨在比较替雷利珠单抗单药和研究者选择的化疗二线治疗不可切除、局部晚期或转移性ESCC患者的疗效和安全性。研究纳入了513例既往接受过系统化疗(不包括抗PD-1/L1抑制剂)的不可切除或转移性ESCC患者,并按1:1比例随机分成试验组(n=256)和对照组(n=256),分别接受替雷利珠单抗单药和研究者选择的化疗。主要终点为总生存期 (OS),次要终点包括客观缓解率(ORR)、反应持续时间(DoR)等。研究结果显示,在意向性治疗人群(ITT)中,与对照组相比,试验组的中位OS显著更长[8.6个月(95%CI:7.5个月-10.4个月)vs6.3个月(95%CI:5.3个月-7.0个月),单侧P=0.0001],风险比(HR)为0.70 (95%CI:0.57-0.85)。此外,试验组和对照组的ORR分别为20.3%和9.8%,中位DoR分别为7.1个月和4.0个月。这表明替雷利珠单抗作为二线治疗,能够为ESCC患者带来显著的生存获益,具有优良的抗肿瘤活性。在安全性方面,与对照组相比,试验组安全性结果更优,治疗相关不良事件(TRAE)(73.3%vs93.8%)和≥3级TRAE的发生率(18.8% vs 55.8%)均更低。其中,最常见的TRAE包括天冬氨酸转氨酶升高(11.4%)、贫血(11.0%)和甲状腺功能减退(10.2%),安全性特征与既往研究一致,未发现新的安全性信号。
七、亿帆医药:升白药Ryzneuta在欧美成功上市
2023年11月17日,亿帆医药发布公告称,其控股子公司亿一生物(Evive Biotech)研发的艾贝格司亭α注射液(内部研发代码:F-627,英文商品名:Ryzueuta,中文商品名:亿立舒)获得美国FDA批准,用于成年非髓性恶性肿瘤患者在接受容易引起发热性中性粒细胞减少症的骨髓抑制性抗癌药物治疗时,降低以发热性中性粒细胞减少症为表现的感染发生率。2024年3月25日,艾贝格司亭α注射液在欧盟上市销售,产品有效期为5年。

艾贝格司亭α注射液是一种重组融合蛋白,其氨基末端包含G-CSF,羧基末端包含人IgG2-Fc片段。该产品通过与G-CSF受体进行特异性结合,可刺激中性粒细胞前体和成熟中性粒细胞的存活、增殖、分化和功能。艾贝格司亭α通过让这些关键的白细胞增殖,增强免疫系统的抗感染能力,从而预防有可能出现的影响治疗结果的化疗剂量减少和延迟问题。该产品具有新型结构,可提供独特且天然长效的治疗方案。
2018年1月,亿一生物完成了艾贝格司亭α注射液首个在美国及欧洲开展的3期临床试验(简称“04试验”),并达到预设主要疗效终点,受试者耐受情况良好,安全性达到预期。2020年1月,亿一生物收到在中国开展的艾贝格司亭α注射液的3期临床试验《统计数据图表合集》,统计结果表明,该产品中国3期临床试验的有效性结果已全面达到临床试验预设评价标准,疗效与对照药品(原研药品重组人粒细胞集落刺激因子)相当。
2020年6月,亿一生物收到在美国及欧洲开展的第二个国际3期临床试验(简称“05试验”)《统计数据图表合集》,结果显示,第二个国际3期临床试验成功达到预设主要疗效终点和次要疗效终点,药物疗效与对照药品相当;2020年7月,公司完成了05试验有关免疫原性的中和抗体检测,结果为阴性,标志着无药物相关的抗体产生;自此,艾贝格司亭α注射液海内外开展的1~3临床试验均达到临床试验预设目标。
值得一提的是,亿一生物早先已经与正大天晴南京顺欣达成合作,后者获得了艾贝格司亭α注射液在中国的独家商业化权益,亿一生物将获得最高可达2.1亿元的首付款与里程碑付款;此外,亿一生物也已经与APOGEPHA公司签订合作协议,后者将获得艾贝格司亭α在德国的独家经销权。在这项合作中,亿一生物将获得首付款40万美元、最高不超过100万美元开发里程碑付款及最高不超过3750万美元的销售里程碑付款。
八、海和药物:首款在日上市的国产新药谷美替尼
2024年6月24日,日本厚生劳动省批准靶向c-MET激酶抑制剂谷美替尼片(研发代号SCC244)在日本上市,用于治疗具有METex14跳变的局部晚期或转移性非小细胞肺癌(NSCLC)。
谷美替尼因其明显的临床疗效与安全耐受的双重优势,已于2023年3月被中国国家药品监督管理局纳入突破性治疗药物品种附条件上市,并被美国食品药品监督管理局(FDA)授予孤儿药资格。系统临床前研究结果显示,谷美替尼体外能够高选择性、强效抑制c-Met激酶酶活,在多种METex14跳变和MET基因扩增的肺癌动物模型上显现出快速诱发肿瘤消退的卓越疗效;谷美替尼成药性好,体内代谢稳定、易透过血脑屏障,且安全耐受。
本次适应症的上市许可申请主要基于SCC244-108关键Ⅱ期研究(GLORY研究,NCT04270591)的有效性和安全性数据。全球多中心临床研究(GLORY研究)显示,患者总体客观缓解率(ORR)为65.8%,其中初治患者ORR高达70.5%,经治患者ORR为60.0%;总体人群中位无进展生存期(mPFS)为8.5个月,总体人群中位总生存期(mOS)为17.3个月,对脑转移患者疗效明确。
总结
欧美日等发达国家市场是所有中国创新药企向往的“神圣之地”,究其原因在于中国与以美国为代表的发达国家医疗支出和创新药定价的差距,在美国等发达国家市场上市是创新药放量的关键。
2021年美国医疗费用支出达4.3万亿美元,中国医疗费用支出达1.1万亿美元,中美医疗费用支出的差距达3.9倍。而中美人均医疗支出差距更是明显,2021年美国人均医疗费用支出达1.3万美元,中国人均医疗费用支出达784美元,中美医疗支出差距达16.6倍。
除了以上获批的8款国产新药相关的研发企业外,还有恒瑞医药、信达生物等知名药企纷纷布局海外市场,期待国产创新药出海再创佳绩。
<END>
想要解锁更多药物研发信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!
















 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论