
































近年来,小核酸药物成为生物制药企业的研发热点,小核酸药物专指靶向作用于RNA或蛋白质的一类寡核苷酸分子,包括反义寡核苷酸(ASO)、siRNA、aptamer等。
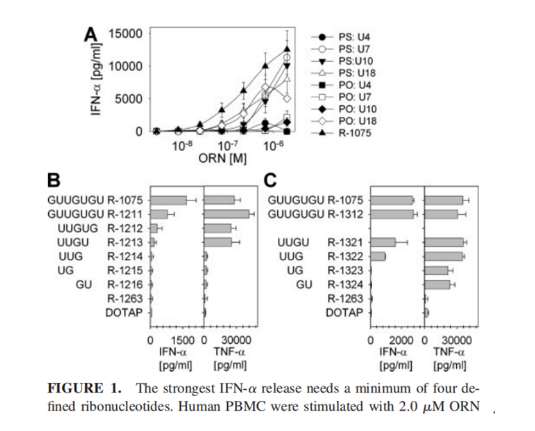
我们都知道,小核酸药物具有特异性强、设计简便、研发周期短、靶点丰富等优势;与此同时,小核酸药物的作用时间与小分子药物相比,更加持久,长能达到半年,短能达一周,这将大大提高患者的依从性,减少患者持续服药的次数;当然,任何药物都会存在一些不良反应,小核酸药物也不例外,下面汇总了部分已上市产品的不良反应,以及部分ASO药物的共同不良反应,汇总发现共同不良反应为注射部位反应、头痛、发热、呼吸道感染、咳嗽、呕吐等;同时,已上市的几款药物存在的主要安全性问题包括:肝毒性、肾毒性和超敏反应。具体见下图1和下表2;

图1 毒性反应[1]
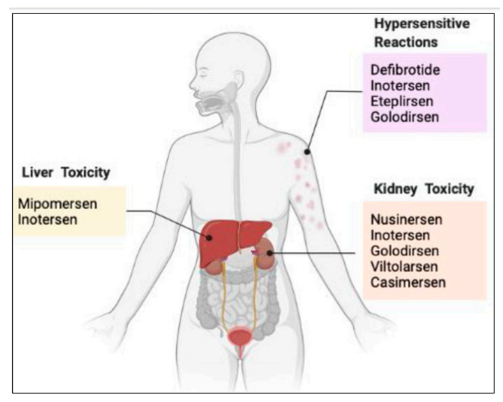
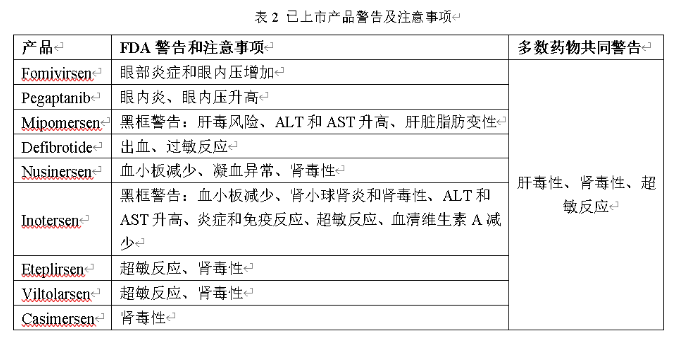
下面我们对产生这些毒性反应来源、影响因素以及非临床安全性研究过程中的关注点进行分析:
毒性反应来源
小核酸药物与小分子药物不同,小核酸药物进入体内后,主要是通过与目标mRNA进行结合,从而达到效果;因此在这里,毒性来源主要有两种:一种是依赖序列杂交,另一种是不依赖序列杂交;而依赖序列杂交存在两种毒性,一种为靶向毒性,另一种为脱靶毒性;而不依赖序列杂质会产生如下反应:
免疫原性、高组织浓度、血小板减少、凝血异常、补体激活等;而器官组织浓度较高又受脱靶毒性和免疫反应的影响;而血小板减少也会受免疫反应的影响。其中靶向毒性、脱靶毒性以及血小板减少主要是受药物的序列影响;而免疫反应和高组织浓度主要是特定机体和药物序列影响。
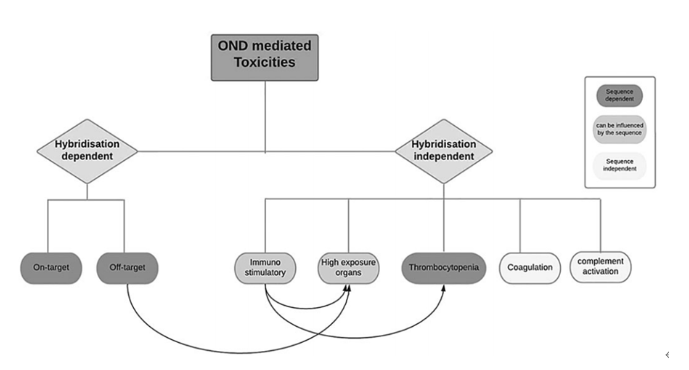
图2 毒性反应来源[2]
靶向毒性
靶向毒性主要是指药物与目标mRNA结合,发挥的疗效过于强大已经超过预期疗效(比如治疗糖尿病出现的低血糖)或者不是预期的靶组织,而造成其他组织的不良反应;虽然目前没有明确的例子,但由于小核酸药物的作用时间,与小分子药物相比,作用时间更长,这就导致其洗脱时间较长;因此OSWG以及日本药品和医疗器械管理局《寡核苷酸治疗产品非临床安全性评价指导原则》[5]建议对其进行评估;与此同时,PMDA 建议,参考 ICH S6 开展评价,对于靶向毒性,可以在具有药理学活性的1种或2种动物中开展非临床安全性评价。当无药理学相关动物种属时,靶向毒性可采用在动物中具有药理学活性的替代物开展毒性研究。 通常只在1个种属中采用替代物开展毒性研究。对于剂量选择,与生物类似药相似,参考ICHS6 的建议高剂量组应产生最大的药理学作用。
脱靶毒性
脱靶毒性是指与靶序列相似的序列杂交而产生的脱靶效应,2009年,迈阿密制药公司OPKO开发用于湿性黄斑变性的siRNA药物bevasiranib在III期临床试验中效果不佳而被终止。此外,由于给药障碍和脱靶毒性等问题久未解决,许多跨国制药公司相继撤离了小核酸药物领域;脱靶毒性通常是比较严重的,比如急性肝毒性;对于脱靶毒性,OSWG建议可以先遵循一些简单的步骤,比如:①采用计算机模型(in silico)筛选出潜在的互补序列(脱靶向序列);②剔除一些序列,比如:在组织中表达很少的;③在主要的靶细胞和脱靶细胞中,进行体外筛选,探索出在靶细胞和脱靶细胞中的最大反应活性的浓度;④体外浓度应该在预期剂量水平以上,这样才能够反应对脱靶效应的潜在毒性。
对于杂交依赖的脱靶毒性,PMDA 建议,考虑到人与动物基因组的差异,建议采用生物信息学的方法和体外考察人源细胞基因表达的方法进行评价;同时,PMDA建议,对于脱靶毒性,寡核苷酸应在啮齿类和非啮齿类动物中开展非临床安全性评价,具体的实验动物选择一般应考虑代谢的种属差异以及同类产品的毒性特点,选择与人体具有相似代谢、同类产品毒性较为敏感的动物。对于脱靶毒性,参考 ICH M3(R2) ,高剂量组设置一般为最大耐受剂量( maximum tolerated dose,MTD) 、暴露饱和剂量、最大可行剂量( maximum feasible dose,MFD) 、50 倍临床暴露量或限制剂量( 1000 mg·kg -1);
不依赖于序列杂交的毒性
免疫原性
一般寡核苷酸与自然界中寡核苷酸具有某些相同的分子特征,可能产生免疫毒性。然而研发过程中通常会对这些化合物进行化学修饰,以提高稳定性、安全性、细胞吸收及疗效。常见的修饰位点包括杂环的碱基,不同基团结合的核酸链,糖或二脂键进行修饰。
这些修饰可能增加免疫反应;根据文献报道,免疫原性反应机制主要是寡核苷酸药物与识别模式受体结合(如:TLRs)后(见下图3),激活先天免疫系统,从而产生免疫反应;据报道,寡核苷酸免疫调节主要受寡核苷酸序列影响、富含鸟嘌呤、尿嘧啶的序列更加容易与TLR7/8结合(见下图4);另外,一些修饰也会促进炎症反应,比如PS修饰,PS修饰就容易产生免疫原性反应;虽然2'F、2'MOE以及LNA修饰,能够减少免疫刺激反应,但由于已上市的产品均存在该不良反应,因此OSWG建议对免疫原性反应进行必要的研究;而PMDA建议,对于免疫毒性试验可以根据重复给药毒性等试验的结果进行评价,则可不进行独立的免疫毒性试验。如适用,参考 ICH S8《人用药物免疫毒性研究》。
图3 免疫反应机制[3]
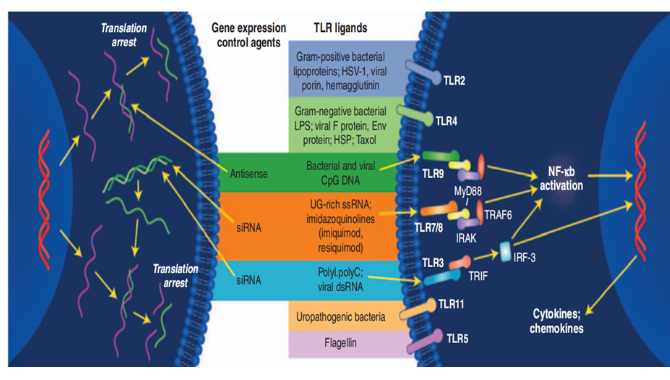
图4含有不同U和G的序列与TLR7/8结合[4]
血小板减少症
血小板减少偶尔在临床前模型中观察到,在volanesorsen、 inotersen和 drisapersen特别是PS-ASO中观察到,与PS-ASO相比,siRNA的产品血小板减少很少被观察到,只有siRNA包裹在脂质体中观察到血小板减少的症状,这可能与阳离子脂质分子本身有关;在PS-ASO患者观察后,发现血小板减少依赖于序列,与物种没有关系,不同物种之间差异不大。Ions公司对102个序列进行评估,在NHP中发现有40%观察到血小板下降,而在人类中,有20%(16条序列)序列观察到血小板下降。
血小板下降的机制依然在研究,PS主干修饰已经被证明是导致血小板下降的重要原因之一,据研究报道,PS-ASO与血小板糖蛋白VI的序列特异性结合,进而激活人类血小板,触发血小板-白细胞聚集物的形成。
器官毒性
全身给药后寡核苷酸药物最高浓度是在肝脏和肾脏,通常我们会认为,肝毒性以及肾毒性主要是因为在肝脏和肾脏蓄积导致,然而,根据文献报道产生肝毒和肾毒的原因与蓄积无关,对ASO的碱基、序列的主干进行微小的修改,会对亲和力以及毒性产生巨大的影响;在很多情况下,在亚啮齿类动物研究中,显微镜观察到肝损伤或坏死,而药物在肝脏中的剂量暴露远小于被接受的肝毒性的剂量。
研究显示,肝毒性很大程度受ASO的化学修饰,碱基主干的修改,与某些类型的ASO蛋白质相互作用有关。在低亲和性的化学修饰(比如2'OMe,MOE)需要很高的剂量组织浓度才能触发临床前物种的肾毒性,采用中亲和性(比如:MOE)和高亲和性(LNA)的化学修饰在人类中,观察到肾毒性。
总结:
综上所述,对于小核酸药物需要特别关注的毒性主要包括靶向毒性、脱靶毒性、器官毒性以及免疫原性;少数药物需要关注血小板减少;虽然已上市的产品均未观察到靶向毒性,但由于其作用时间较长,因此还是需要进行研究。
以上是基于当前认知水平进行分析总结,如有错误,欢迎大家留言指正。
参考文献:
[1]Feryal Alhamadani等人,Adverse Drug Reactions and Toxicity of the Food and DrugAdministration–Approved Antisense Oligonucleotide Drugs;Drug Metab Dispos 50:879–887, June 2022;org/10.1124/dmd.121.000418
[2]Aure´ lie Goyenvalle等人,Considerations in the Preclinical Assessment of the Safety of Antisense Oligonucleotides;NUCLEIC ACID THERAPEUTICS Volume 33, Number 1, 2023 Mary Ann Liebert, Inc. DOI: 10.1089/nat.2022.0061;
[3]Sudhir Agrawal & Ekambar R Kandimalla;Antisense and siRNA as agonists of Toll-like receptors;Published online 6 December; doi:10.1038/nbt1042;等等
<END>
要解锁更多企业药品研发信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药品各国上市情况、药品批文信息、销售情况与各维度分析、市场竞争格局、一致性评价情况、集采中标情况、药企申报审批信息、最新动态与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论