































近年来,全球制药巨头纷纷涌入罕见病市场。2018年,武田斥资650亿美元收购夏尔,此后,罗氏以43亿美元的对价收购Spark Therapeutics;阿斯利康更豪掷390亿美元收购Alexion;葛兰素史克以19亿美元收购罕见癌症靶向治疗公司Sierra;辉瑞以116亿美元收购Biohaven公司、以54亿美元收购GBT公司。罕见病龙头不愧是跨国巨头竞相追逐的并购对象,巨头们正在罕见病领域用并购手段加码布局。
其中,已经有26年历史的罕见病制药企业Biomarin频频与罗氏、安进、吉利德等多个巨头传出绯闻。这家美国药企,曾连续多年霸榜美国GEN杂志所评选的生物制药领域TOP10并购名单,是十大热门标的之一。

药融云数据显示:二十多年前,医学界对罕见病的研究以及罕见病的治疗都是十分稀少的。罕见病较小的市场规模、和巨大的研发难度,都让医药公司对这一领域望而却步。BioMarin以治疗粘多糖贮积症I(或MPS I)的药物Aldurazyme为起点,创立了BioMarin并在同年推动了该药的临床试验,最终在2003年Aldurazyme获得了批准,成为了首个获批的用于治疗粘多糖贮积症I的酶替代疗法。
如今,BioMarin已经拥有了8款商业化产品。对于中国市场来说,最熟悉的或许是Vimizim。这款用于治疗IVA型黏多糖贮积症(MPS IVA)的药物,从2008年启动立项启动到最终2014年首次正式获批上市,整个过程用了7年时间。2019年,Vimizim获得国家药品监督管理局批准,成为中国首个黏多糖贮积症药物。
此外,该公司还拥有多条潜力管线,比如血友病的基因疗法Valoctocogene Roxaparvovec、儿童软骨发育不全症药物Vosoritide等等,是罕见市场中管线布局很深远、成熟的药企。这种出色的研发能力,使其经常被许多小型药企视作行业标杆。目前,BioMarin开发的血友病A基因疗法Valoctocogene Roxaparvovec在行业内遥遥领先,颇具看点,很有可能是该公司走向下一个成功的“胜券”。
(BioMarin研发管线-截至2023年2月27日)
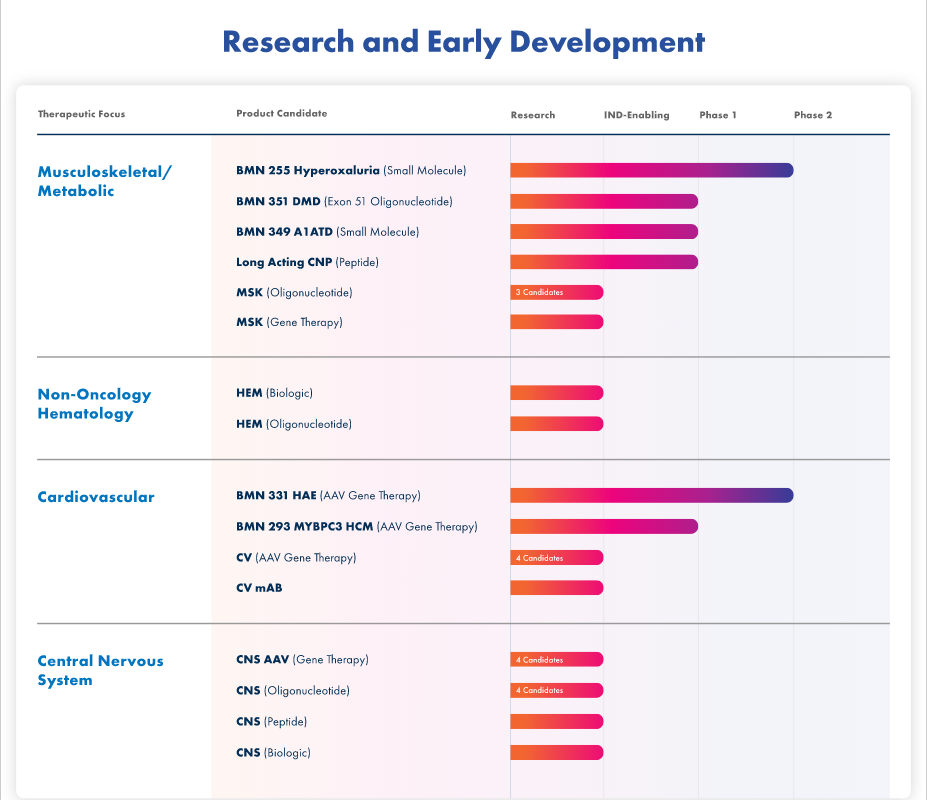
NO.1 关于基因疗法Valoctocogene Roxaparvovec
在2022年第三季度,欧盟委员会(EC)有条件地批准了Roctavian(Valoctocogene Roxaparvovec),未在2022年进行销售,Roctavian有可能成为世界上第一款治疗A型血友病的基因治疗药物,值得期待。
2022年8月24日,欧盟批准Roctavian(Valoctocogene roxaparvovec)基因疗法的有条件上市,用于治疗体内不含凝血因子VIII抑制物和腺相关病毒5(AAV5)抗体的重度A型血友病成人患者。2023年3月6日,BioMarin公告,FDA确定提交正在进行的ROCTAVIAN第3期GENEr8-1研究的三年数据分析构成了一项重大修订,因为有大量额外数据,并将新的PDUFA目标行动日期定为2023年6月30日。第三阶段研究包括134名参与者,是迄今为止最大的血友病基因治疗研究。
去年底,FDA批准了全球首个针对重度和中重度血友病B的基因疗法——Hemgenix(etranacogene dezaparvovec)。Hemgenix的标价为每次使用350万美元,创下了最昂贵的一次性基因疗法的新纪录。Roctavian,适用于严重的血友病A,价格预计约为250万美元。
罕见病的在体基因治疗策略大致可分为两类:针对由于特定基因缺陷引起的疾病,可通过递送缺失功能基因的正常版本来纠正,简称“基因回补”,针对由于错误折叠的毒性蛋白引起的疾病,可以通过递送基因编辑工具对突变基因进行敲除或纠正,简称“基因编辑”。
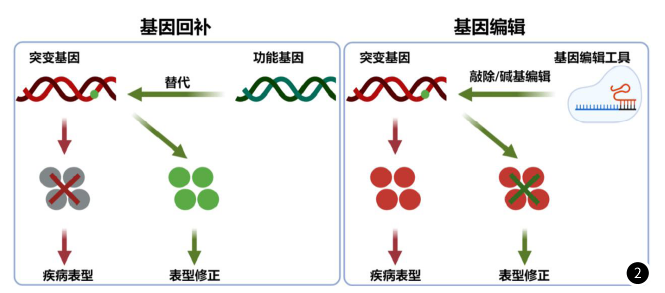
罕见病虽然罕见,但患病总人数众多,多数缺乏有效治疗方法。基因治疗作为一种新兴的治疗手段,有望解决单基因病的治疗困境。而AAV作为目前最安全、长效的病毒载体,已经被应用于多项罕见遗传病的临床试验,涵盖眼病、血液病、神经系统疾病等多个领域。
根据迄今为止的临床经验来看,AAV载体是治疗罕见单基因病的一个很好的平台。与小分子、抗体和蛋白质替代疗法相比,基因疗法作为一个平台还处于发展的早期阶段,仍旧有很多问题需要解决,如免疫原性的问题。但是基因治疗是可以实现一次性治疗疾病的方法,对罕见病治疗的发展有着十分深远的影响。
NO.2 靠“7+1”款商业化产品支撑起商业大厦
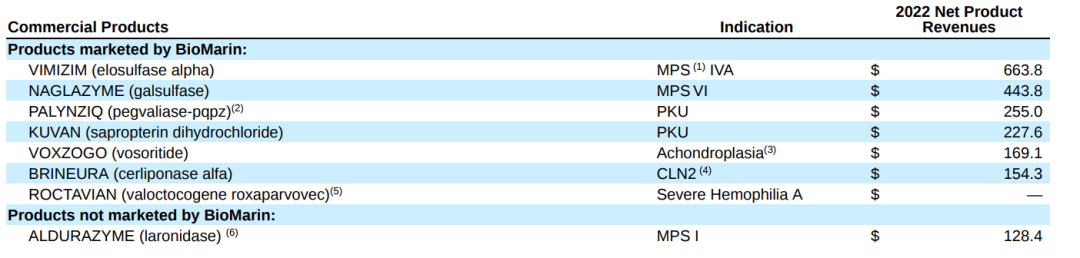
(注:欧盟委员会(EC)在2022年第三季度有条件地批准了Roctavian,但在2022年没有销售;ALDURAZYME(laronidase)由赛诺菲(Sanofi)销售。)
目前,BioMarin已经拥有了Vimizim、Naglazyme、Kuvan、Palynziq、Brineura、Voxzogo、Aldurazyme等多个治疗罕见病如PKU、CLN2、MPS的药物,以及多个以AAV为载体的基因治疗药的研发管线。在2022财年,Vimizim产品净销售额最高,其次是Naglazyme。
1.Vimizim(elosulfase alfa)
Vimizim是一种用于治疗MPS IVA(一种溶酶体贮积症)的酶替代疗法。MPS IVA是一种以N-乙酰半乳糖胺-6-硫酸酯酶(GalNS)活性缺乏为特征的疾病,导致溶酶体过度储存某些称为糖胺聚糖(GAGs)的复合碳水化合物,如硫酸角质素和硫酸软骨素。这种过度储存会导致全身性骨骼发育不良、身材矮小和关节异常,从而限制活动能力和耐力;胸部畸形会损害呼吸功能,颈部关节松动会导致脊柱不稳和潜在的脊髓压迫。其他症状可能包括听力丧失、角膜混浊和心脏病,最初的症状往往在生命的头五年变得明显。Vimizim已获准在美国、欧盟和其他国际市场上市。
Vimizim药物研发信息(部分,完整内容请登录“药融云数据库www.pharnexcloud.com/?mh”查看)
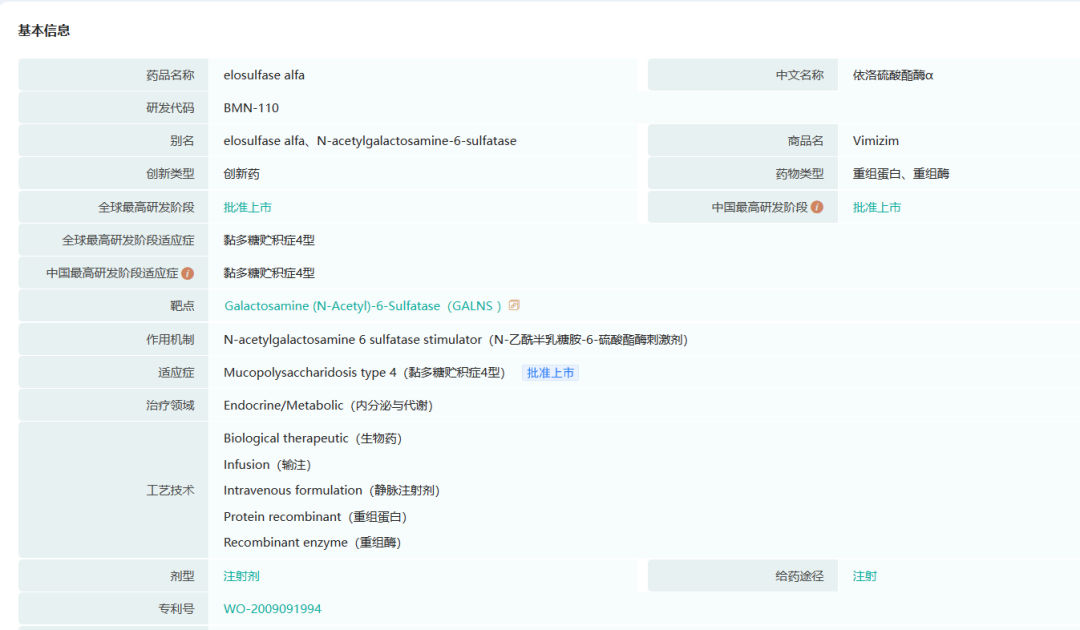
截图来源:药融云全球药物研发数据库
2.Naglazyme(galsulfase)
Naglazyme是N-乙酰半乳糖胺4-硫酸酯酶(芳基硫酸酯酶B)的重组形式,适用于MPS VI患者。MPS VI是一种使人衰弱、危及生命的遗传性疾病,目前尚无其他药物治疗,其病因是缺乏芳基硫酸酯酶B,这是一种分解GAG所需的酶。患有MPS VI的患者通常会逐渐恶化,并经历多种严重和虚弱的症状,这是由于体内组织中碳水化合物残留物的积累所致。这些症状包括:生长抑制、脊髓压迫、肝脾肿大、关节畸形和活动范围减小、骨骼畸形、心血管功能受损、上呼吸道阻塞、肺功能降低、频繁耳部和肺部感染、听力和视力受损、睡眠呼吸暂停、不适和耐力下降。Naglazyme已获准在美国、欧盟和其他国际市场上市。
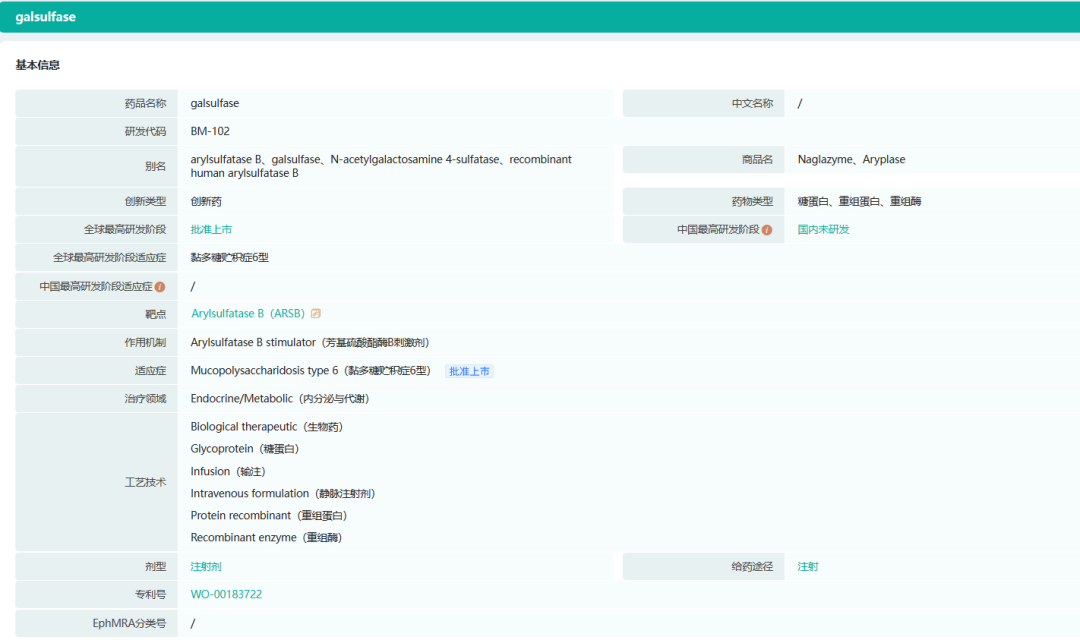
截图来源:药融云全球药物研发数据库
3.Palynziq(pegvaliase-pqpz)
Palynziq是一种聚乙二醇化重组苯丙氨酸(Phe)氨裂解酶,通过皮下注射降低血液中Phe的浓度。Palynziq是BioMarin受批准的第二种治疗苯丙酮尿症的药物。Palynziq被批准在美国上市,用于患有苯丙酮尿症(PKU)的成年患者,这些患者的血液苯丙氨酸(Phe)浓度在现有管理下超过600微摩尔/升。Palynziq也被批准在欧盟和澳大利亚上市,用于16岁及以上的患者。
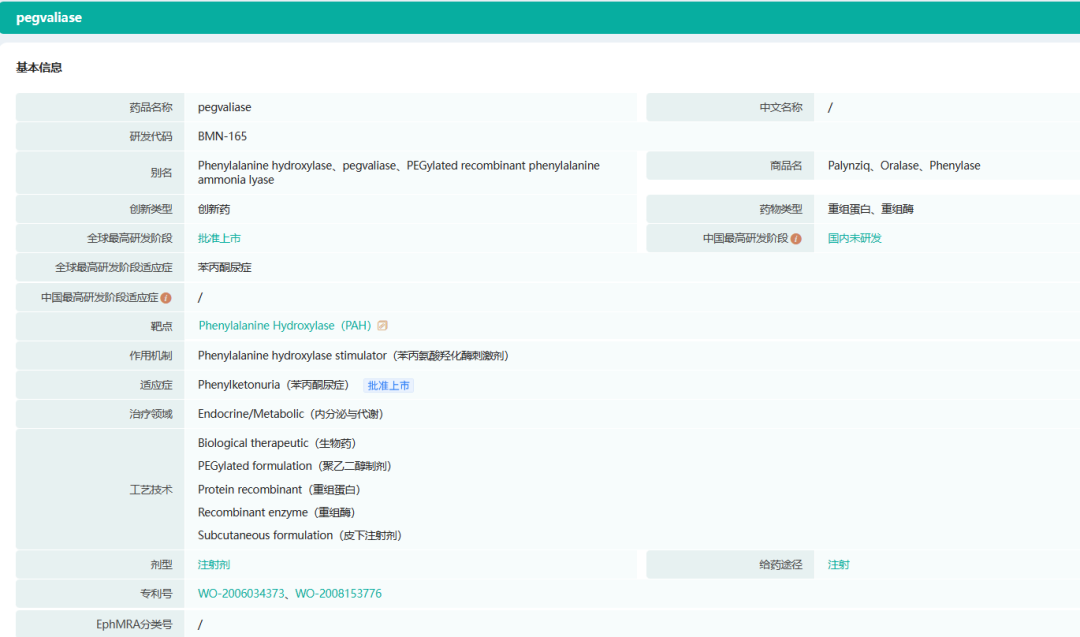
截图来源:药融云全球药物研发数据库
4.KUVAN(sapropterin dihydrochloride)
Kuvan是6R-BH4的专有合成口服形式,6R-BH4是苯丙氨酸羟化酶(PAH)的天然酶辅因子,适用于PKU患者。Kuvan是第一种治疗苯丙酮尿症的药物,苯丙酮尿症是一种遗传性代谢疾病,在发达国家至少有5万名40岁以下的确诊患者。大约30%至50%的苯丙酮尿症患者可以从Kuvan治疗中获益。苯丙酮尿症(PKU)是由苯丙氨酸(Phe)代谢所需的一种酶(PAH)活性缺乏引起的。苯丙氨酸是一种必需氨基酸,存在于所有含蛋白质的食物中。如果没有足够数量或活性的PAH,PHE会在血液中积累到异常高的水平,导致各种严重的神经系统并发症,包括严重的精神发育迟滞和脑损伤、精神疾病、癫痫发作和其他认知问题。
由于20世纪60年代和70年代初实施的新生儿筛查工作,发达国家几乎所有40岁以下的苯丙酮尿症患者都在出生时被确诊。目前,苯丙酮尿症可以通过限制苯丙氨酸的饮食来控制,并辅以营养替代产品,如配方奶粉和特制食品。然而,大多数患者很难坚持严格的饮食以达到充分控制血液苯丙氨酸水平所需的程度。
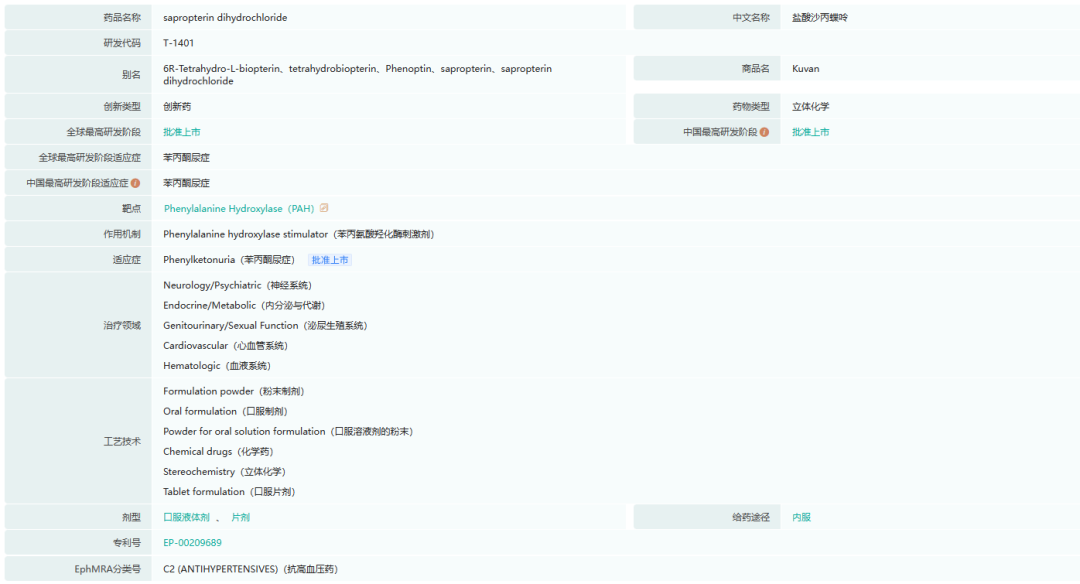
截图来源:药融云全球药物研发数据库
5.VOXZOGO(vosoritide)
Voxzogo是一种每日注射一次的C型利钠肽(CNP)类似物,用于治疗软骨发育不全。在软骨发育不全患者中,由于成纤维细胞生长因子受体3基因(FGFR3)的功能获得突变,软骨内骨生长(骨组织形成的基本过程)受到负调控。Voxzogo作为FGFR3下游信号通路的正调节因子,促进软骨内骨生长。
Voxzogo已获准在欧盟、澳大利亚和巴西上市,用于两岁及以上患有开放性生长板软骨发育不全的患者,并在日本上市,用于所有年龄患有开放性生长板软骨发育不全的儿童。FDA加速批准了VOXZOGO在美国用于5岁及以上患有开放生长板的软骨发育不全患者,并授予BioMarin与Voxzogo的加速批准有关的罕见儿科疾病优先审查凭证(PRV)。2022年第一季度,BioMarin将此PRV以1.1亿美元的价格出售。
BioMarin仍在继续进行VOXZOGO对软骨病儿童的安全性和有效性的研究。2022年2月23日,BioMarin宣布了VOXZOGO在患有软骨症的5岁以下婴幼儿中进行的2期随机、双盲、安慰剂对照临床试验的结果,2022年6月13日,又宣布了分析的其他细节。52周的结果显示,与安慰剂相比,VOXZOGO在身高(按年龄和性别调整)和年化生长速度方面有优势,这与之前在五岁以上儿童中治疗一年后观察到的改善一致,并且没有明显影响上下身段比。安全性方面与3期研究和产品标签人群中的大龄儿童基本一致。与接受VOXZOGO治疗的儿童(7%)相比,安慰剂组的严重不良事件(SAEs)更高(18%)。所有的SAEs,包括一个致命的呼吸停止(报告为一个接受治疗的婴儿的婴儿猝死综合症,之前就有呼吸道疾病),都被研究者认为与治疗无关。最常见的不良事件是轻微的、自限性的注射部位反应。2023年1月3日,BioMarin宣布提交了II型变异申请,EMA随后验证该II型变异申请,以扩大VOXZOGO在欧盟的适应症,治疗2岁以下的软骨病儿童,此外,还向FDA提交了补充新药申请(NDA),治疗5岁以下的软骨病儿童。2022年8月3日,BioMarin宣布VOXZOGO用于治疗有枕骨大孔压迫风险的两岁以下婴儿的干预性2期研究完成了入组。该研究正在调查VOXZOGO对有可能需要手术的婴儿的安全性,以减轻枕骨大孔的压迫。目前,BioMarin正在评估VOXZOGO作为软骨症以外的其他遗传性身材矮小疾病儿童的潜在治疗方法。由华盛顿特区国家儿童医院赞助的一项为期52周的研究,旨在调查VOXZOGO对部分遗传性身材矮小儿童的治疗效果,该研究正在进行,预计将于2023年完成。12名受试者6个月的初步结果显示,所有亚组都有积极反应,但个体间存在差异。
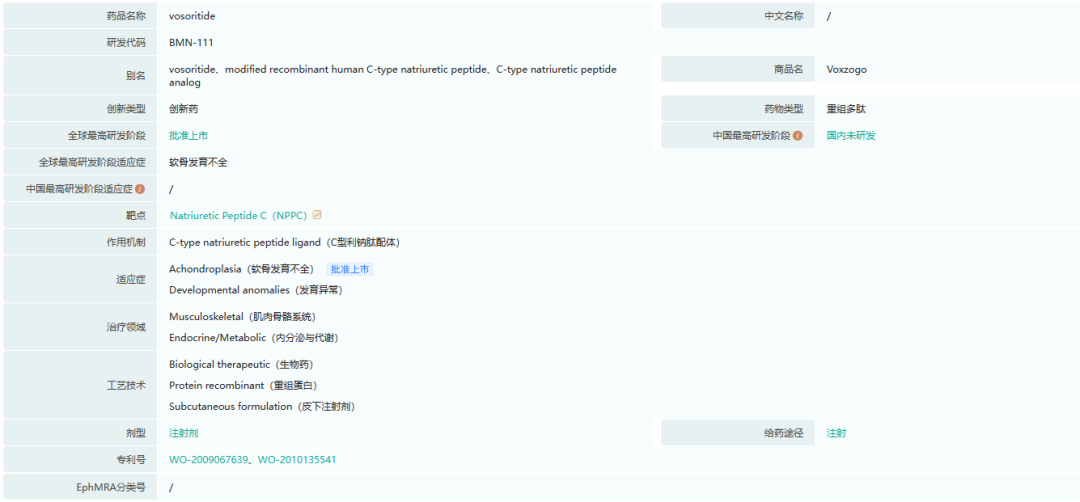
截图来源:药融云全球药物研发数据库
6.BRINEURA(cerliponase alfa)
Brineura是一种重组人三肽基肽酶1(TPP1),用于治疗CLN2(一种Batten病)患者。CLN2是一种不可治愈、进展迅速的疾病,通常导致患者在10-12岁时死亡。患者最初是健康的,但在大约三岁时开始衰退,BioMarin估计全世界有多达1,200至1,600个病例。Brineura是第一个被批准用于减缓患有CLN2疾病的儿童丧失步行能力的治疗方法,也是欧盟首批通过加速审查程序的治疗方法之一。Brineura通过脑室内(ICV)输注给药,并旨在与递送装置(如注射器或其他递送系统)联合使用。Brineura获准在美国(适用于三岁及以上儿童)、欧盟(适用于从出生起的所有年龄段)和其他国际市场销售。
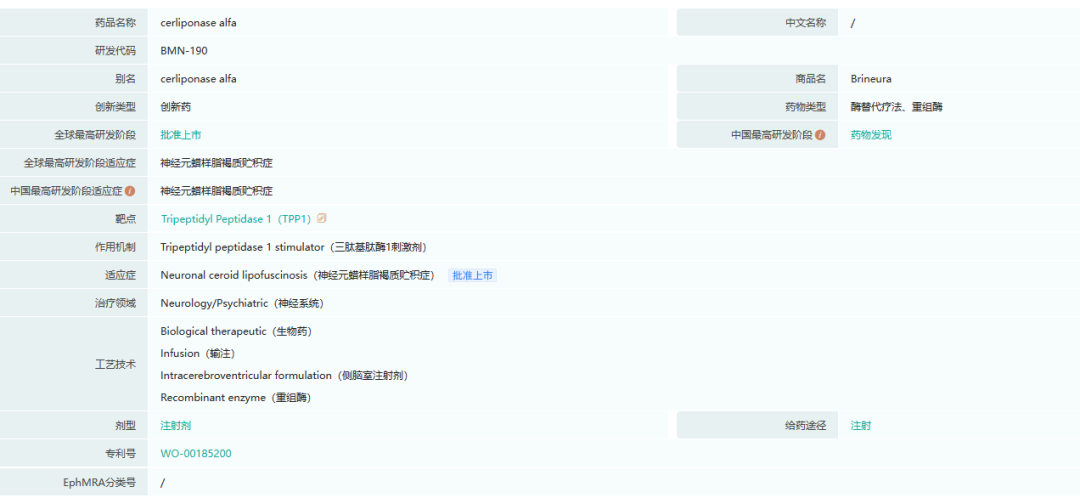
截图来源:药融云全球药物研发数据库
7.ROCTAVIAN(valoctocogene roxaparvovec)
Roctavian是一种腺相关病毒(AAV5)载体基因疗法,旨在恢复严重血友病A患者的凝血因子VIII血浆浓度。血友病A,也称为凝血因子VIII缺乏症或典型血友病,是一种由凝血因子VIII(一种凝血蛋白)缺失或缺陷引起的遗传性疾病。根据世界血友病联盟(World Federation of Hemophilia)对血友病A严重程度的分级,因子VIII活性水平的正常范围在50%至150%之间,以血液中正常因子活性的百分比表示,轻度血友病A的因子VIII活性水平范围在5%至40%之间,中度血友病A的因子VIII活性水平范围为1%至5%,因子VIII活性水平的严重血友病范围小于1%。患有血友病A的人不能有效地形成血凝块,并有因轻微受伤而出血过多的风险,可能危及他们的生命。患有严重血友病的人通常会自发出血到肌肉或关节中。
2023年1月8日,BioMarin公布了Roctavian治疗成人严重血友病A的全球3期研究的三年分析结果。所有132名研究参与者均接受了单剂量的Roctavian治疗,并进行了至少36个月的随访。在一项非干预性前瞻性基线观察研究中,预先指定的112名参与者(其中两人在三年随访前退出研究)在服用Roctavian(滚动人群)后,在第三年,平均ABR从基线降低了80%,治疗出血的平均ABR为1.0(中位数0.0)。对于滚动人群,在第三年,平均年化因子VIII输注率从基线降低了94%,平均年化输注率为8.4(中位数0.0)。截至三年数据截断期,92%的转期人群参与者仍未接受因子VIII预防性治疗。在输注Roctavian三年后结束时,132名参与者的平均因子VIII活性水平为18.8(中位数8.4)IU/DL,通过发色底物(CS)分析测量。在17名参与者(其中一人在达到四年随访前退出研究)中,在数据削减前至少四年给药,在第三年结束时,通过CS测定,平均因子VIII活性为15.2(中位数7.4)IU/DL。该亚人群第四年治疗出血的平均ABR为0.8(中位数0.0),平均年输注率为11.1(中位数0.0)。
除了上述正在进行的Roctavian的1/2期和3期研究外,BioMarin正在进行一项3期、单组、开放标签研究,以评估Roctavian(剂量为6E13 VG/kg)与预防性皮质类固醇联合治疗严重血友病A患者的疗效和安全性;此外,还在进行一项正在进行的1/2期研究,在已有AAV5抗体的严重血友病A患者中使用6E13 VG/kg剂量的Roctavian,以及在有活性或既往因子VIII抑制剂的严重血友病A患者中使用6E13 VG/kg剂量的Roctavian。
8.ALDURAZYME(laronidase)
Aldurazyme是一种高度纯化的蛋白质,其被设计为与人类酶α-L-艾杜糖苷酶的天然存在形式相同,α-L-艾杜糖苷酶是一种通常分解GAG所需的溶酶体酶。MPS I是一种由α-L-艾杜糖苷酶缺乏引起的渐进性、致弱性、威胁生命的遗传性疾病,目前尚无其他药物治疗。患有MPS I的患者通常会逐渐恶化,并经历多种严重和虚弱的症状,这是由于体内所有组织中碳水化合物残留的累积所致。这些症状包括:生长抑制、智力发育延迟和退化(病情严重时)、肝脾肿大、关节畸形和活动范围缩小、心血管功能受损、上呼吸道阻塞、肺功能下降、频繁的耳部和肺部感染、听力和视力受损、睡眠呼吸暂停、不适和耐力下降。
BioMarin通过与赛诺菲(Sanofi)合作开发了该药物。根据与赛诺菲的合作协议,BioMarin负责生产Aldurazyme并将其供应给赛诺菲,收取赛诺菲全球Aldurazyme净销售额的39.5%至50%的付款。Aldurazyme已获准在美国、欧盟和其他国际市场上市。
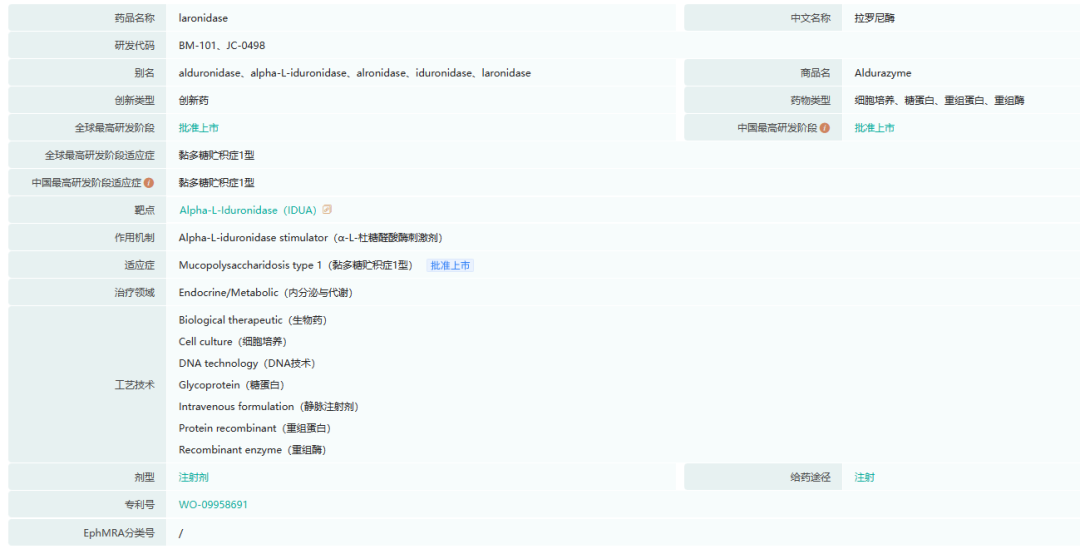
截图来源:药融云全球药物研发数据库
NO.3 财务状况
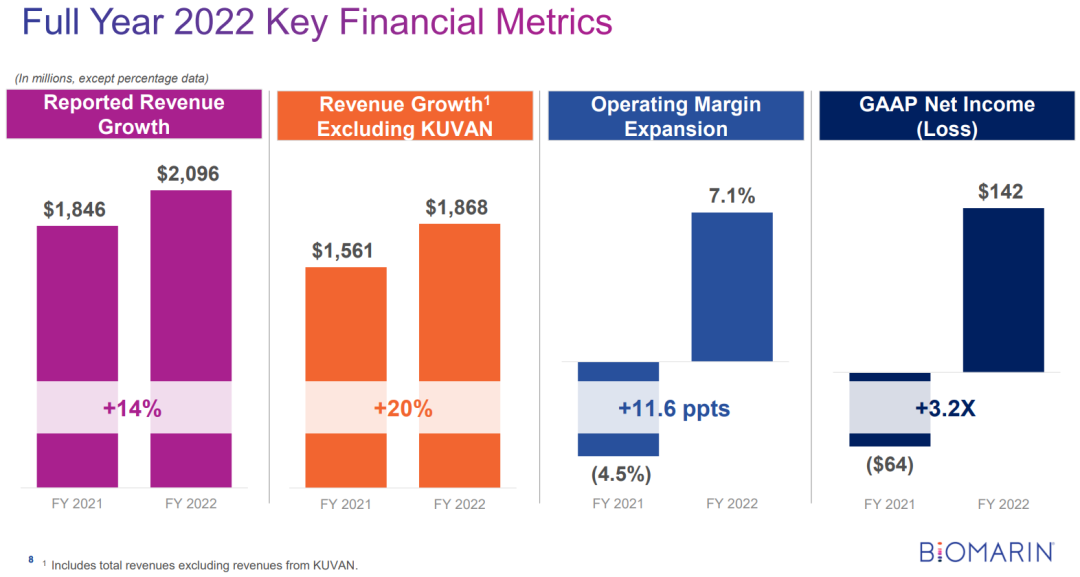
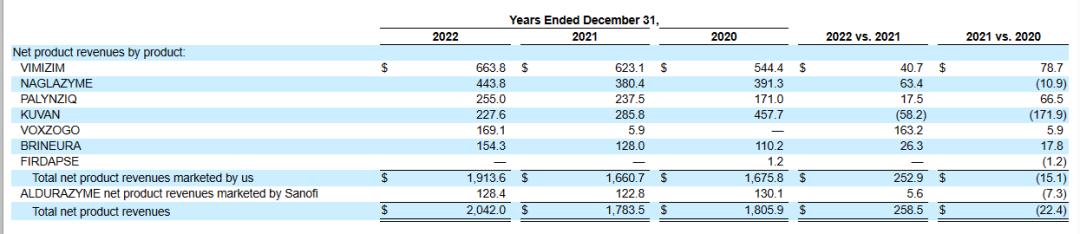
(BioMarin2022年年度报告)
2023年2月28日,Biomarin公布2022年财报,2022全年营收20.96亿美元,同比去年的18.46亿美元增长了14%;研发费用支出6.5亿美元,同比增长3%;净利润1.42亿美元,扭亏为盈。具体产品而言,IV型黏多糖贮积症药物Vimizim销售额6.64亿美元;VI型黏多糖贮积症药物Naglazyme销售额4.44亿美元,增长了17%;苯丙酮尿症药物Palynziq销售额2.55亿美元;沙丙蝶呤(科望)销售额2.28亿美元。
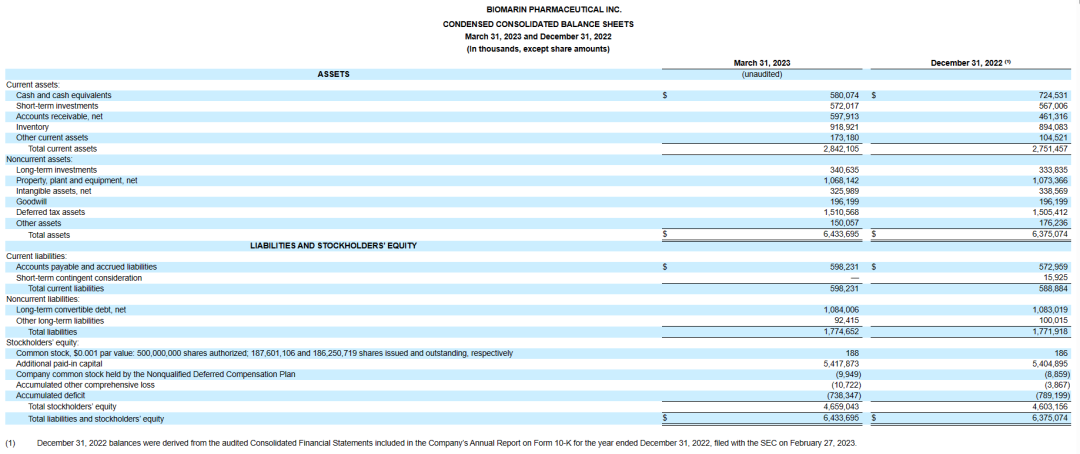
(BioMarin2023年一季度报告)

(2023第一季度BioMarin产品营收表现)
据BioMarin在2023年4月29日公布的2023年第一季度财报,该期间公司营收额约5.96亿美元,研发投入金额约1.72亿美元,公司综合所得约4399.7万美元。截至2023年第一季度末,公司拥有现金及现金等价物5.8亿美元,现有流动资产28.4亿美元。
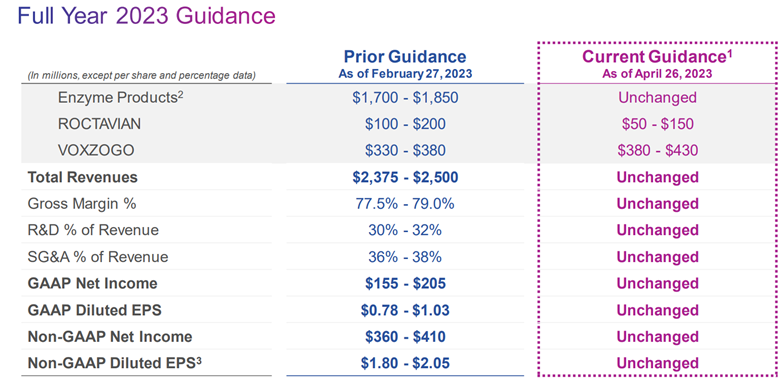
2023年4月26日,BioMarin更新了2023年产品预计营收:酶相关产品:17亿美元~18.5亿美元;Roctavian:5千万美元~1.5亿美元;Voxzogo:3.8亿美元~4.3亿美元。2023全年指引总营收23.75亿美元~25亿美元。
截至2023年6月16日,BioMarin当日股价96.29美元/股,市值约180.74亿美元。

参考:
NMPA/CDE;
药融云数据,vip.pharnexcloud.com/?mh;
FDA/EMA/PMDA;
相关公司公开披露(除标注外,正文图片均来自企业官方);
https://www.biomarin.com
https://investors.BioMarin.com/events-presentations
https://mp.weixin.qq.com/s/TsggydRV3kSxMVdPmnVPnQ
https://mp.weixin.qq.com/s/u6Cd_WTZAKFXz-3U-MzScw
https://finance.sina.com.cn/stock/med/2023-03-10/doc-imykkqpu9783284.shtml
https://mp.weixin.qq.com/s/7bSwzWk_A1GIZTc_D49xHA
想要解锁更多药企信息吗?查询药融云数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药企公司基本信息、投融资情况、产品管线分布、药物销售情况与各维度分析、药物研发情况、年度报告、最新进展动态、临床试验信息、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!

<END>
















 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论