































引言
作为认知最广泛的神经退行性疾病,阿尔茨海默症一直没有行之有效的治疗药物,6月3日,一篇发表在nature neuroscience上的文章揭示了阿尔茨海默症(Alzheimer's disease,AD)最可能接近真相的病因,溶酶体酸化障碍,但是,这和最终开发阿尔茨海默症的药物还有不小的距离。
AD大脑神经病理学改变的特征包括,Aβ蛋白沉积形成的淀粉样蛋白斑,神经原纤维缠结(过度磷酸化tau蛋白组成的胞内聚集体),突触的丧失、萎缩、神经递质系统(例如乙酰胆碱)的选择性耗竭以及路易体(少数情况),以上这些情况导致了神经元之间的信息交流障碍甚至神经元的死亡,最终导致AD。因此目前的药物开发思路主要是围绕清除β样淀粉蛋白(Aβ蛋白),调节Tau蛋白,以及ACHE(乙酰胆碱酯酶抑制剂)等这些热门的领域,但是近年来也有一些新的AD机制和治疗方法兴起,比如炎症理论,干细胞疗法和基因疗法。
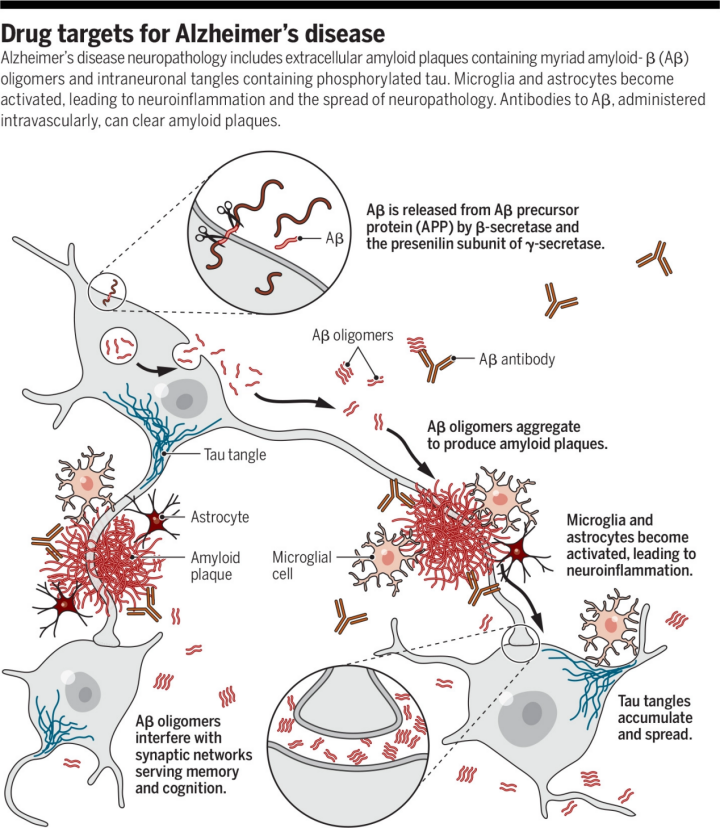
阿尔茨海默病神经病理学包括含有无数淀粉样蛋白-β (Aβ) 寡聚体的细胞外淀粉样蛋白斑块和含有磷酸化tau的神经元内缠结。小胶质细胞和星形胶质细胞被激活,导致神经炎症和神经病理学的扩散。
Aβ蛋白级联假说
Aβ蛋白是弥漫性和神经炎性斑块的主要蛋白质成分,其起源于淀粉样前体蛋白(APP)的蛋白水解。APP是一种1型整合跨膜蛋白,其结构中Aβ的C端部分嵌入细胞膜内。
产生Aβ蛋白需要2个连续的蛋白水解步骤,先由β-分泌酶在Aβ序列的N末端区域切割APP,产生稍短的可溶性N末端(APPsβ)和淀粉样蛋白C末端片段(C99),β-分泌酶对C99的切割释放出APP的C端50个残基,称为APP胞内结构域(AICD)和Aβ。
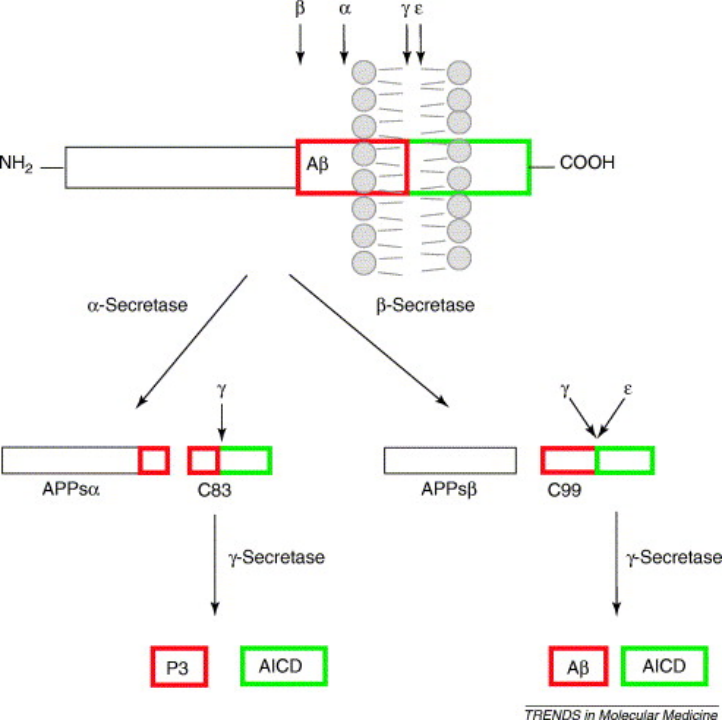
而在其分解过程中,细胞内的溶酶体承担了大部分的工作。溶酶体的酸化障碍导致细胞错误产生的Aβ蛋白不能正常被分解,进而撑破溶酶体,使得细胞破裂将Aβ蛋白释放到细胞外,进一步形成斑块。
Aβ蛋白可以引发一系列的信号级联反应,研究发现,其可以通过以下几条途径降低突触可塑性(突触连接强度可调节的特性(神经递质释放、细胞接受突触的敏感型等))或减少突触的密度。
- 1、与细胞朊病毒蛋白(PrPC)结合,激活Fyn激酶,然后通过NMDA型谷氨酸受体(NMDAR)途径开启对突触的长期抑制作用(LTD)。
- 2、非NMDAR途径,与PrPC和代谢型谷氨酸5受体(mGlu5Rs)形成三元复合物,导致突触可塑性受损和突触的密度下降
- 3、Aβ蛋白的积聚可间接性导致Tau蛋白在大脑区域中的累积和扩散。
- 4、Aβ蛋白可以抑制乙酰胆碱受体(ACHR),诱导LTD,导致突触传导抑制。
药物研发现状
利用单克隆抗体结合细胞外的Aβ蛋白单体/可溶性的聚集体(目前最主流的方法),防止其聚合或者激发下游信号通路。代表性的药物有百健的aducanumab,礼来的donanemab和罗氏的crenezumab。
但根据Aβ信号级联理论,在Aβ蛋白的产生,以及降解的过程,都有机会进行药物的研发。目前一些药物研发是针对APP产生Aβ蛋白过程中的β-分泌酶,研发其抑制剂(Beta secretase inhibitor/BACE),例如法兰克福大学的MH-84,默沙东的MBI-10,不过这些目前都处于临床前的研究阶段,距离成药还有一段不小的距离。
也有一些其他的研究方向,比如
- Aβ聚集抑制剂(曲米酸、鲨肌醇、PBT2)
- Aβ抗原(AN-1792、vanutide、AD02、CAD-106)
- 抗Aβ多克隆抗体(免疫球蛋白)
- γ-分泌酶抑制剂(begacestat、semagacestat和avagacestat)
- γ-分泌酶调节剂(tarenflurbil)
- β-位点淀粉样前体蛋白裂解酶(BACE)抑制剂(LY2811376、LY2886721、AZD3839、verubecestat、atabecestat和lanabecestat)。
阻断其下游通路也可作为AD药物研发的新思路,比如今年6月1日发表在science上的一篇文章,揭示了mGlu5Rs的无声(SAM)变构调节剂(BMS-984923,百时美施贵宝)可以逆转阿尔茨海默小鼠的突触丢失。

也有针对NMDAR途径研发的NMDA药物,虽然这种药物对改善神经认知的效果不错,但是容易引发抑郁。
Tau蛋白假说
Tau是稳定神经元微管的微管相关蛋白(MAP)之一,主要出现在轴突中(与体树突MAP2相比)。
神经元之间的信息传递依赖于微管作为轨道,而tau蛋白与微管相结合以维持其稳定性。当tau的关键位点磷酸化( 主要是Ser262或Ser214),Tau则从已结合的微管中释放出来,导致微管破裂和tau聚集成成对的螺旋丝(PHF)。Tau高磷酸化和神经纤维缠结是AD病理学的关键组成部分,被认为由人类大脑的上游Aβ突触病理学驱动,并与Aβ协同增效以进一步的突触丢失。
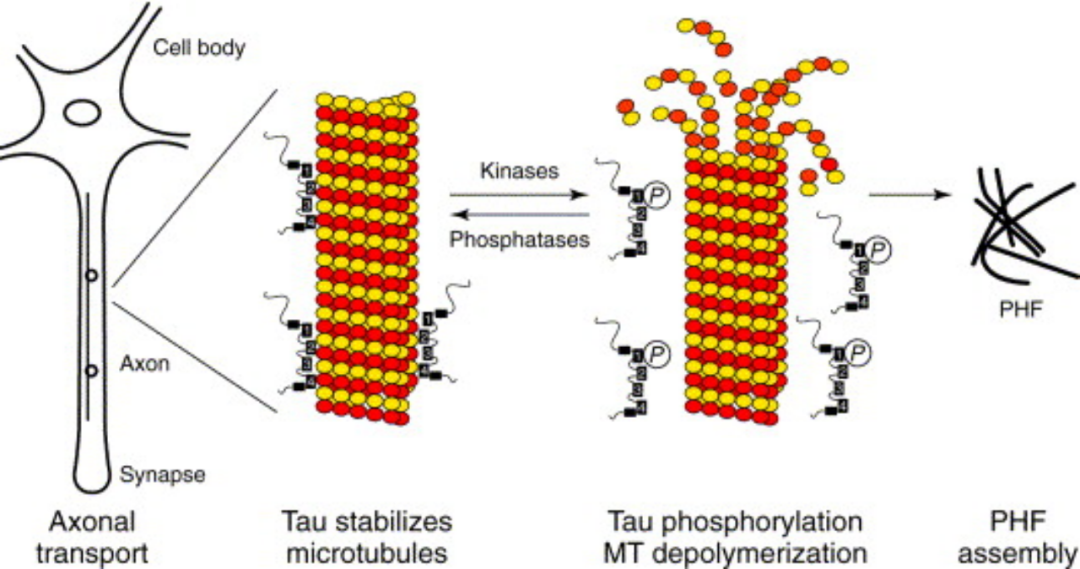
关键磷酸化位点
Tau包含一个酸性N端结构域、一个碱性和富含脯氨酸的中间结构域、一个包含三个或四个内部重复的碱性结构域和一个C端结构域。它可以在多个位点被磷酸化,其中一些调节其微管结合特性。
出现在内部重复序列两侧的两个区域中的几个Ser-Pro或Thr-Pro基序,对tau-微管相互作用只有中等影响,但可用作AD样tau磷酸化的诊断工具的。并且是脯氨酸定向激酶的靶标,例如糖原合酶激酶3、细胞周期蛋白依赖性激酶Cdk5或MAP激酶。 其他位点包括蛋白激酶A(例如Ser214)、微管亲和调节激酶(MARK,在KXGS基序处,包括Ser262、Ser356)或Ca 2+ /钙调蛋白依赖性蛋白激酶(Ser416)的靶点。
tau蛋白物研发思路
许多异常磷酸化位点位于Ser-Pro或Thr-Pro基序上,因此针对AD-tau研发的各种抗体则是与这些磷酸化位点发生反应。
免疫学说
最近,全基因组关联研究表明,神经炎症的积累是介导AD开始和进展的遗传危险因素。在人类研究中,超过25个遗传位点与发生AD的风险有关,其中大多数主要在小胶质细胞中表达并与神经炎症有关。神经炎症可以促进Aβ蛋白的产生并且诱导tau的磷酸化。
神经炎症及其下游通路
Aβ斑块周围的小胶质被激活到促炎症状态,并分泌白细胞介素(IL)-1β。IL-1β促进神经元中可溶性淀粉样蛋白前体蛋白(sAPP)α的产生,sAPPα通过激活核因子kappa B(NF-κB)信号传导小胶质细胞中Pro-IL-1β的产生。同时,Aβ激活NLRP3炎症体,从灭活的Procaspase-1中产生激活的Caspase-1,导致小胶质细胞进一步分泌IL-1β。该循环使神经炎症事件成为慢性,并通过激活p38有丝分裂原激活蛋白激酶(p38-MAPK)途径诱导神经元中tau的高磷酸化和突触蛋白的减少。
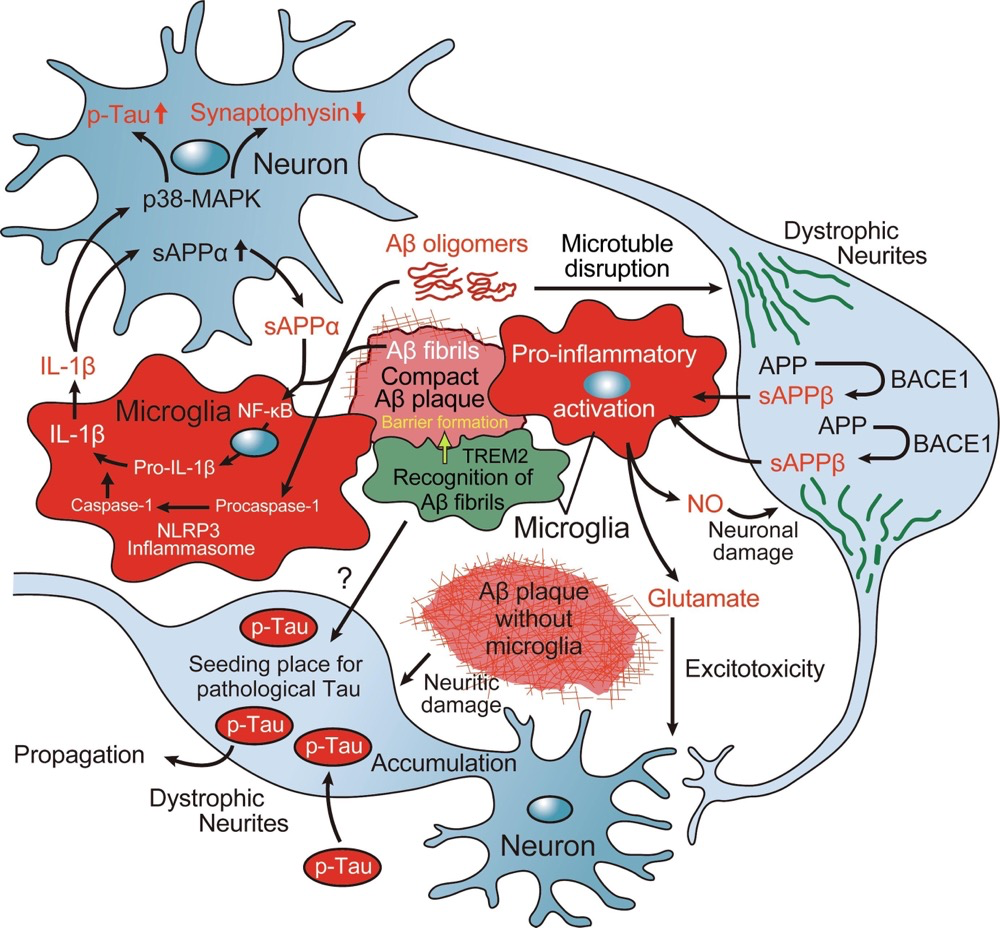
神经病理学研究假设的小胶质细胞在阿尔茨海默病(AD)中的代表性促炎症
调节神经炎症的因子
与AD神经炎症机制有关的调节因子有,骨髓细胞-2(TREM2)跨膜蛋白(其水解裂解的减少会加剧神经炎症,是大脑微胶质活动的调节剂),富含核苷酸结合域的亮氨酸重复序列(NLR)和含有pyrin结构域3(NLRP3),含有半胱天冬酶募集结构域(ASC)的凋亡相关斑点样蛋白(ASC)、CD33 和 CD22 。
钙稳态调节剂
在大脑中,小胶质细胞是最丰富的免疫细胞类型,占所有免疫细胞的80%以上。而钙稳态与小胶质细胞活化密切相关,Aβ增加细胞内钙水平,这反过来又有助于小胶质细胞中NLRP3炎症小体的活化。钙稳态调节剂家族蛋白(Calhm,Calhm1,Calhm2和Calhm3)的作用在AD研究领域受到越来越多的关注。在敲除Calhm2的小鼠中,其Aβ沉淀和神经炎症都显著降低,并减轻了AD相关的认知障碍。
基因疗法
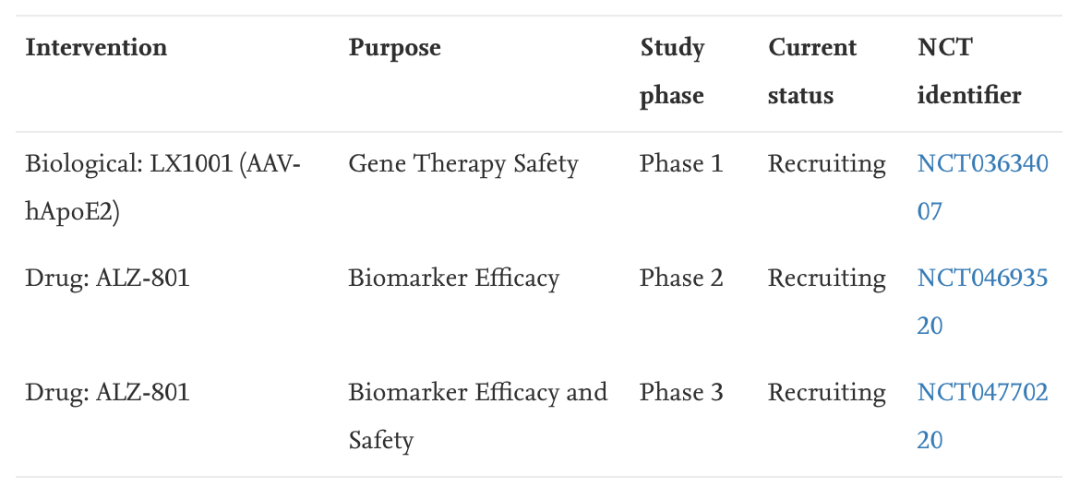
ApoE4
目前已经明确的和AD有关的基因是ApoE基因突变,特别是ApoE4基因。目前也有一些临床试验针对此基因展开。
过氧化物酶体增殖剂激活受体γ共激活剂-1α(PGC-1α)
PGC-1α主要参与调节β-APP切割酶1(BACE-1)的生产,该酶负责Aβ的生产。一项涉及接触hPGC-1α的APP23转基因小鼠的临床试验表明,小鼠的记忆力有所改善,淀粉样沉积物也有所减少。此外,由于NGF和脑源性神经营养因子的表达增加,还具有神经保护作用
CRISPR/Cas9
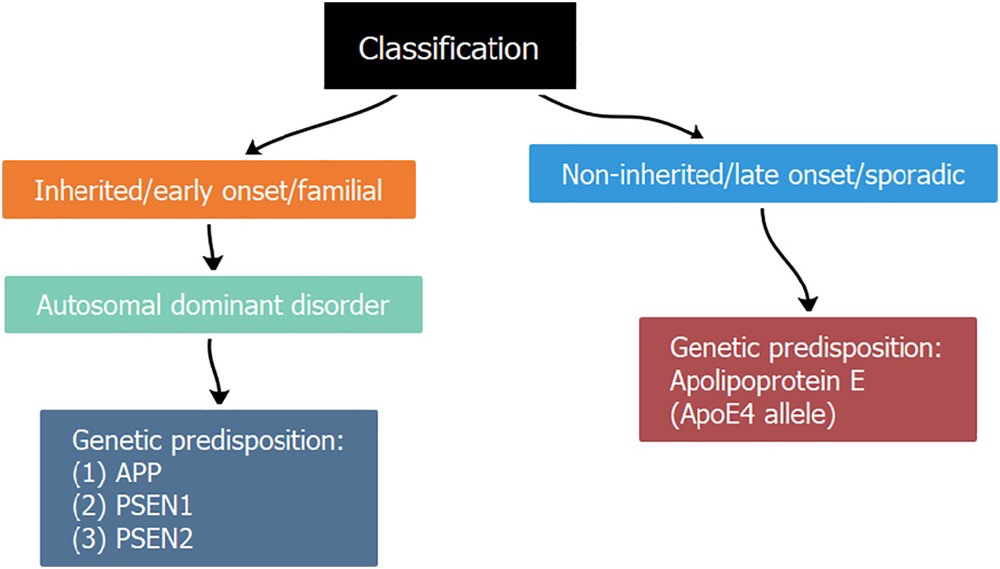
AD具有APP、PSEN1和PSEN2基因突变形式的易感性的遗传基础,它还与ApoE4等位基因的表达有关,这些基因位点可以作为治疗的靶点。
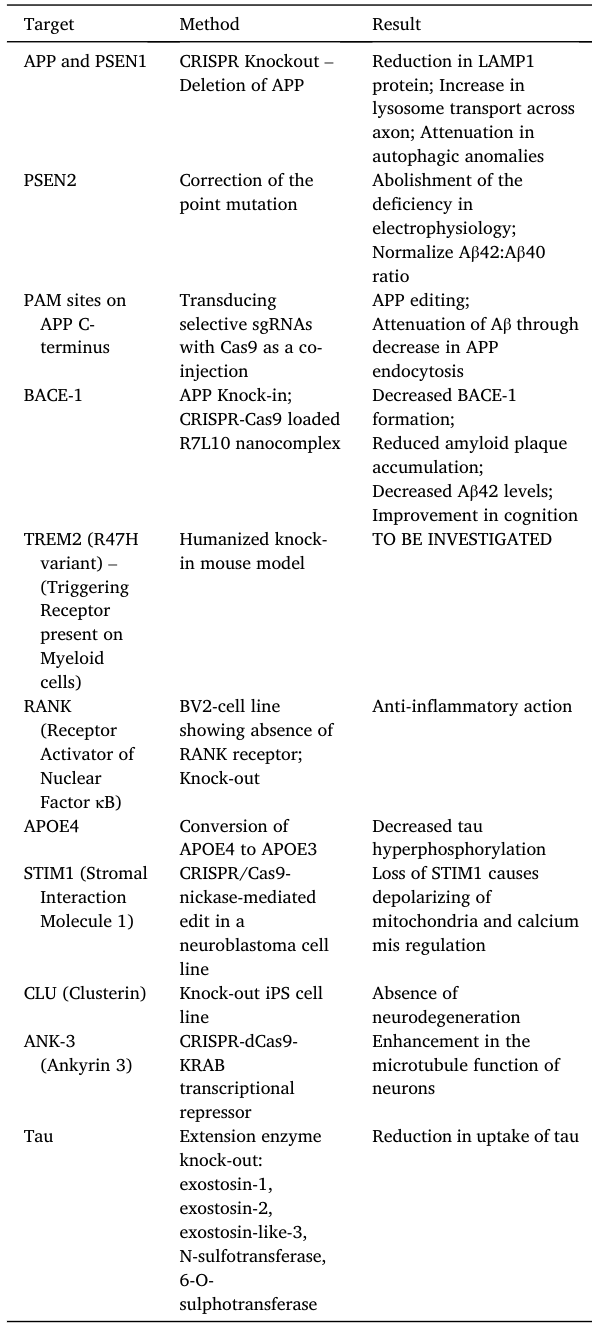
目前已经有一些利用CRISPR的靶点治疗研究,具体如下:
小结
越来越多的证据表明,AD 是一种异质性疾病,由超出典型教条的各种病理生理机制引起。例如,多达三分之一的临床诊断为 AD 的患者没有 Aβ 积累,而许多在死后活检中诊断为 AD 的患者并没有表现出认知障碍 。现今的理论认为,阿尔茨海默症可能和癌症一样,拥有不同的致病成因,因此识别AD的分子生物标志物以区分不同亚型,可能是开发出更有效的药物的关键,而未来差异化病因的阿尔兹海默症的治疗药物将会百花齐放。
参考来源:
[1] Alzheimer's disease: Aβ,tau and synapticdys function
[2] Tau in Alzheimer's disease
[3] Treatments for Alzheimer's disease emerge
[4] Reversal of synapse loss in Alzheimer mouse models by targeting mGluR5 to prevent synaptic tagging by C1Q
[5] Microglial Calhm2 regulates neuroinflammation and contributes to Alzheimer’s disease pathology
[6] Sustained Trem2 stabilization accelerates microglia heterogeneity and Aβ pathology in a mouse model of Alzheimer’s disease
[7] Roles of microglia in Alzheimer’s disease and impact of new findings on microglial heterogeneity as a target for therapeutic intervention
[8] An update on Alzheimer's disease: Immunotherapeutic agents, stem cell therapy and gene editing
[9] Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets
推荐阅读:
<END>
















 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论