





























高效液相色谱法(HPLC)是药物开发和质量控制过程的重要检测手段。本文阐述了高效液相色谱法开发的一般流程。
1 概述
高效液相色谱法开发是理论和实践的结合。在掌握高效液相色谱法的相关基础知识的同时,了解高效液相色谱法系统中的各个变量对分析结果的影响才能更好地选择色谱条件。基于已有信息和经验,根据化合物的理化性质,并根据试验数据逐渐优化色谱条件,才能最终确定合适的高效液相色谱法检测。
高效液相色谱法开发的步骤应考虑如下内容:
a) 信息调研:通过文献或其他来源,收集原料药的相关信息
b) 溶解度研究:确定溶解度情况和最大吸收波长
c) 色谱条件选择:根据溶解度研究结果、化合物的保留时间等信息确定色谱柱、流动性、进样样品
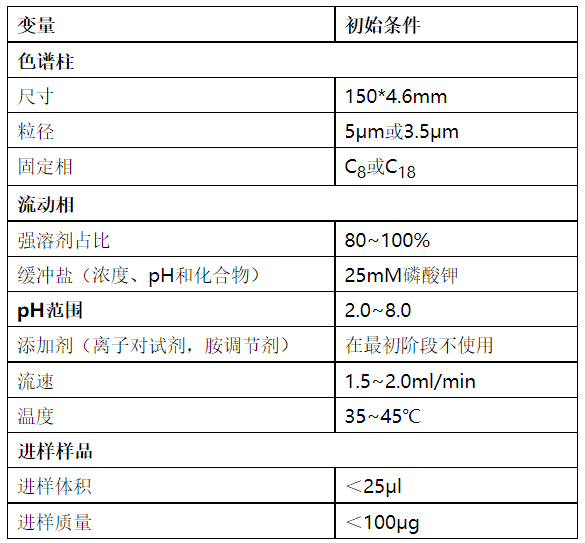
d) 进行初步的高效液相色谱法分析检测,最初阶段的高效液相色谱法测试倾向采用的参数见表1。
e) 确定梯度或等度模式
f) 优化色谱条件提高分离度:通过改变流动性组成、缓冲盐、pH、色谱柱、流速、温度等实现合适的分离度
g) 进行原料药的强降解试验确定方法的适用性
h) 确定系统适用性参数
i) 方法总结并完成开发报告
j) 进行方法验证
表1.最初阶段的高效液相色谱法分析测试倾向选择的参数
2 方法开发过程中考虑要点
一个完整的高效液相色谱法检测方法,应当包括样品配制,分析检测和最终的图谱分析全过程的必要信息,而不只是与色谱条件相关的信息。然而,色谱条件的确定却是方法开发最为重要的内容。
2.1 信息调研
全面收集相关化合物的信息能够为高效液相色谱法开发提供更好支撑。尤其是很多仿制药开发而言,化合物的公开信息众多。一般的信息来源途径包括审评文件、公开文献和仿制药手册。甚至在某些时候,能够直接找到已有的检测方法。对于全新的化合物,以化合物类型搜索相关的支撑信息往往也能够缩短方法开发的时间。例如,多肽类,苯环上位置异构等在方法开发时存在一些通用性质的理论和经验。
2.2 溶解度研究
高效液相色谱法开发过程中的溶解度研究一般是评估原料药在在水、缓冲液、氢氧化钠、甲醇、乙腈、氯仿、己烷、四氢呋喃等多种水和有机溶剂中的溶解度。需要注意的是,由于样品检测过程中可能存在温度波动,除去考察室温环境下的溶解情况,也需要关注不同温度下的溶解情况。一般而言,原料药在所选稀释剂中应具有良好的溶解性(最好为1mg/ml),根据溶解度选定合适的溶剂。在所选溶剂中进一步进行光谱扫描,一般选择200-400nm范围,以确定药物的光谱特征。
2.3 样品稀释剂(溶剂)的选择
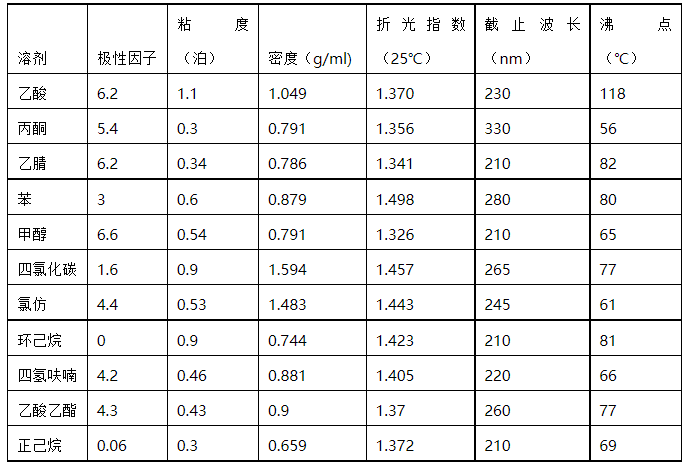
样品所选用的稀释剂(溶剂)应适合所选的方法及预期的目的。常见的溶解的物理性质见表1。对其主要要求如下:
a) 能够溶解主要分析物,稳定性良好
b) 不干扰分析物响应
c) 防止分析物与容器表面的相互作用
表1. 常见溶剂的物理性质
确定好溶剂之后,需要进一步明确合适的样品制备方法。对于制剂样品常常需要开发针对主要分析物的提取方法。但是一般而言,方法开发早期都是仅仅针对化合物进行的,在方法确定之后可以根据需要进一步进行方法优化或补充。
2.4 波长选择
波长选择是方法开发的关键步骤。为了选择波长,应采用所选溶剂制备合适浓度的溶液,并在紫外分光光度计上扫描。在紫外扫描的基础上,将供试品溶液注入配备光电二极管阵列检测器的高效液相色谱法系统,并采集光谱。一般情况下,应当选择能够最佳响应的波长用于药物分析。但在某些时候,出于某些原因,例如避免干扰,也可以选择其他波长。
2.5 色谱系统
正向色谱和反向色谱是目前最为常用的两种色谱,根据不同的化合物性质可以选择合适的色谱系统。
2.5.1 正相色谱法
正相色谱的固定性极性大于流动相,一般情况下极性越强的化合物与固定相的相互作用越强,保留时间越长。如果出现以下情况,则首选正相色谱法:
a) 样品溶解在非极性溶剂中,如己烷、氯仿、二氯甲基乙烷等(如果使用反相柱,直接进样可能存在问题)。
b) 反相色谱柱对样品的保留能力得太强,即使是100%的有机相,50-60分钟内仍然没有峰。
c) 反相色谱柱对样品没有保留能力。
d) 在分离过程中,反相色谱法无法获得足够的峰分离度。
e) 样品由位置异构体、非对映异构体和立体异构体组成。
f) 对于在水相中会分解的化合物,正向色谱很有用。
正向色谱可以分析脂质、糖、甾体、异构体和多核芳烃等。对于正相HPLC,初始阶段的尝试实验可使用100%强溶剂,例如采用氰基柱(250 * 4.5μm,5μ)时,可以采用100%的异丙醇作为流动性,可以洗脱分离确定出所有0.5<K<20的成分。同样地,也可以采用100%IPA的流动性以评估是否需要进行梯度洗脱。为了改变强溶剂的选择性,可以用氯甲烷、MTBE(甲基-1-丁基醚)、乙腈或乙酸乙酯等替代异丙醇。但是,溶剂的选择往往取决于检测器类型。
对于正向色谱,一些常用的固定相按照分离效果递减的顺序为氰基>二氧化硅>二醇>氨基等,可以根据需求进行选择。对于碱性和酸性化合物,可以在流动相中添加三乙胺和乙酸,以防止拖尾。
2.5.2 反相色谱法
高效液相色谱法的首选应该是反相色谱法,化合物的分离取决于具有不同程度亲疏水性的固体相对溶质分子的可逆解吸/吸附。其固定性极性小于流动相,一般情况下极性越高的化合物保留时间越短。根据原料药的性质(酸性/碱性/中性)选择适合的条件,可以采用合适的梯度洗脱方式进行高效液相色谱法检测的首次尝试。为了进一步优化色谱条件,应当对首次检测的色谱图进行仔细分析。根据分析结果,可以通过改变流动相组成、缓冲液pH值、柱填料、柱温度、流速等,以获得最佳分离条件。确立了初步的色谱条件后,应当再多次进样分析,经过多次检测确认可重复性,最终获得适当的分离条件,再进行进一步的条件优化。
2.5.3 等度或梯度模式的选择
液相色谱检测中,若流动性的组成比例保持不变,称为等度洗脱,若流动性的比例逐渐改变,则称为梯度洗脱。梯度洗脱可能会缩短分析周期、提高分离能力。
等度模式和梯度模式的设计的条件基本相同。在初始阶段,应当是一段空白梯度,这可以用来检查基线噪声。在末尾阶段应当是一段空白梯度,与初始的流动性比例一致。
为了确定样品应当采用梯度还是等度,可以参考参考文献【1】中的方法。首先采用5~100%乙腈-缓冲液进行梯度洗脱,流速为2.0 ml/min,持续60 min,据此估算强溶剂的最佳初始和最终百分比。可以重复运行几次以确认分离的再现性。计算总梯度运行时间与第一个和最后一个组分的梯度时间之差之间的比率。如果比率为˂0.25,则应选择等度模式,如果比率˃0.25,选择梯度模式则更好。
2.5.4 反相离子对色谱法
如果反相色谱中不能实现足够的分离度,则使用离子对试剂是非常有用的。RP-HPLC和离子配对RP-HPLC的唯一区别是在流动相中加入离子对试剂,以提高离子样品的选择性。然而,除非有明确的支撑信息,应当首先反向色谱,然后才是反向离子对色谱。流动相的有机溶剂会影响离子对试剂的溶解度,最好使用甲醇,而不是乙腈和四氢呋喃。在反相离子对色谱中,缓冲液的浓度应为25mm左右。流动相pH值与离子对直接相关,离子对带负电还是带正电,取决于分析液是碱还是酸。对于碱或阳离子样品,可选择戊烷/己烷或更高碳氢化合物磺酸盐离子对试剂,对于酸或阴离子样品,可使用四乙基氢氧化铵作为离子对试剂。
2.6 方法开发参数的优化
2.6.1 缓冲溶液的选择
特定pH值的流动相可实现酸性或碱性成分的有效分离。缓冲盐溶液应无紫外吸收,或缓冲盐溶液的紫外吸收波长应小于有机溶剂的波长。此外,还应考虑其他条件,如稳定性、溶解度和与分析物的反应性。缓冲溶液的容量可以用pH值、缓冲液的组成和缓冲液的浓度来评估。pH值等于缓冲液的pKa可提供最佳缓冲容量。大多数缓冲液都有足够的缓冲容量,可将流动相pH值控制在pKa±1左右。C8-或C18-键合硅胶基固定相高效液相色谱法柱通常用于反相色谱,一般而言,它们在2~8的pH范围之外,稳定性较差。因此,应当选择可以将pH值控制在2~8范围内的缓冲盐体系。当然有些色谱柱的pH耐受范围更宽或更窄,遵照其说明书使用即可。
2.6.2 缓冲液pH值的选择
对于不同pKa的化合物,其保留时间随流动相的pH改变而改变。酸性化合物的保留时间随pH降低而延长,碱性化合物则相反。为确保化合物处于100%非离子状态,流动性的pH值至少高于或低于pKa 1.5个pH单位,这有利于实现分离。较低的pH值(1–4)有利于减小峰拖尾和提高的耐用性。相比之下,中等pH范围(4–8)则能提高选择性和提供更好的保留时间。表2中列出了用于高效液相色谱法分离的不同缓冲液。
表2.高效液相色谱法中常用的缓冲盐

2.6.3 流动相选择
流动相组成的优化是高效液相色谱分析的关键部分。一般情况,所选流动性沸点应高于柱温20~50℃,但低于100℃,粘度不大于5*10-4Pa s。在所有的有机溶剂中,乙腈和甲醇是RP-HPLC中最为常用的有机溶剂。接下来是四氢呋喃,但四氢呋喃流动相易被氧化,其使用范围较小。在实际的优化过程中,通常需要采用不同有机相和不同pH值的缓冲溶液作为流动相进行试验,根据结果确定最终的流行相以实现样品中各个组分的最佳分离。对于反向色谱,流动相中有机相的比例增加,保留时间会缩短,但可能导致峰的分离程度不够。如果在流动相中使用100%有机溶剂后没有分离,则应降低溶剂强度以获得适当的保留。在多组分分离(许多物质/分子/混合物)且分辨率较弱的情况下,应尝试不同极性指数的有机溶剂,甚至两种或两种以上有机溶剂的组合,以获得有效分离。溶剂选择可以进一步参见参考文献【2】中的内容。
2.6.4 色谱柱选择
色谱柱是高效液相色谱系统的重要组成部分,在分析中起着至关重要的作用。为高效液相色谱法选择色谱柱时应考虑的参数包括:柱填料、颗粒的大小和形状、柱的长度和直径、碳负荷百分比、孔隙体积、端盖。对于反相色谱,可使用多种色谱柱,如C8、C18、氰基类–CN和氨基类–NH2等。没有两种色谱柱完全相同,不同供应商的色谱柱可能在上述参数方面有所差异。不用色谱柱的主要差异是键合相载体的表面积,因为较大的表面积赋予较大的保留能力(C18>C8>C3>C1)。为了确定方法中的色谱柱类型,应使用不同色谱柱采用不同的色谱柱进行实验,以获得最佳分离效果。根据实验数据,选择的色谱柱应能将主峰与所有已知杂质分离,并能适应流动相的变化。色谱柱的类型多样,根据不同的化合物性质可以进行选择,进一步参见参考文献【3】中的内容。
2.6.5 柱温选择
通常,最好在温和的温度环境下优化色谱条件,但是如果在室温下,选择不同的色谱柱或不同流动相的任何组合也无法实现峰对称性,则可以考虑采用高于环境温度的柱温度。如果柱温升高,峰对称性和峰保留时间降低,但温度变化会改变离子化化合物的pH值和pKa值,可能会实现离子化合物更好地分离。
2.6.6 试验浓度和注射量的选择
试验浓度的选择取决于API在选定波长下的响应。通常进样的化合物量应当小于100μg。如果样品需要进行配制,需要明确配制过程中分析物没有损失。通常,建议进样量为10~20μl,但根据需求(例如样品浓度过低,样品响应过低),可将进样量增加至50–100μl。在选择高进样量之前,应确保在选择较高进样量时,色谱柱不会过载,且分辨率和峰对称性不会受到影响。
2.6.7 过滤器相容性
如果样品在进样前需要采用滤膜过滤,应当选择合适的滤膜,并检查滤膜对药物的吸附情况。首先,所需的滤膜应当与样品的溶剂相容性良好。在进行吸附性考察时,在稀释剂中制备样品和标准溶液,并使用几种种不同类型的滤膜进行过滤。过滤标准溶液的结果应与未过滤标准溶液的结果进行比较,过滤部分的结果应在未过滤标准溶液和离心/未过滤样品溶液的±2.0%范围内。
3 总结
高效液相色谱法的方法开发是一个渐进的过程,并伴随药物开发过程不断优化,同时根据不同的分析需求,参数也可能进行调整。通常,在完成初步的方法开发后,可以考虑进行强降解试验确检测方法的分离效果和适用性。伴随原料药工艺的优化、制剂处方工艺的变化,稳定性考察的新增杂质等因素,高效液相色谱法也需要进行相应的调整。
参考文献:
(1)纷繁,等度洗脱和梯度洗脱的选择及条件计,https://www.bzwz.com/bbs/topic_1628_1.html;
(2)万事皆空,高效液相色谱溶剂的选择,药事纵横;
(3)药事一刻,高效液相色谱柱的选择;药事纵横;
(4)MukeshMaithani, Development of Novel Stability Indicating Methods Using LiquidChromatography, 1.6 General Considerations for HPLC Method development.
相关阅读:《HPLC:“三板斧”在低限度杂质方法开发中的应用》
<END>













 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论