
































三、线性及范围、重复性(精密度中的重复性)
线性:系指在设计的范围内,线性试验结果与试样中被测物质浓度呈比例关系的能力。设计线性实验是为了保证,在一定范围的浓度进样,为了保证你的样品浓度与测得的峰面积或者峰高成正比关系。如果在线性范围内,你的样品浓度与峰面积不成正比,那么直接就说明了你的检测方法是存在问题的。
具体做法:称取对照品精密稀释,制备一系列对照品溶液进行测定,至少制备5各不同浓度水平。(注:有关物质测定时,各杂质必要的浓度点为50%、100%、150%,具体可以设置为20%、50%、80%、100%、150%、200%,当然也可以设置其他的浓度点,请各位自行设计,这里只是举例。含量测定时,必要的浓度点为80%、100%、120%,具体设计点包含这3各必要点后,可以自行设计。)
线性20%处的浓度该如何确定?线性20%处的浓度需至少包含该杂质定量限,而100%处的浓度为杂质的限度浓度及该杂质在该样品中的最大限度浓度(限度浓度主要是根据杂质的性质来确定,如果一般杂质暂定为主成分检测浓度的0.1%),例如某杂质C的定量限为0.205µg/ml,我在实验时为了方便一般设计杂质C线性的在20%处的浓度就为0.205µg/ml,那么在50%、80%、100%、150%、200%线性点处的杂质C 的浓度就分别为0.5125µg/ml、0.820µg/ml、1.025µg/ml、1.5375µg/ml、2.050µg/ml。
做杂质线性实验,目的就是为了证明,我们我采取的检测方法,在一定范围浓度进样条件下,保证各杂质能够被准确检测出来。(举个栗子:某项目中主成分的进样浓度为1.0mg/mL,而杂质C为一般杂质,其限定浓度定为0.1%,那么杂质C的最大进样浓度就为1.0µg/ml,而以上述杂质C定量限为例,还定为0.205µg/mL,那么在20%、50%、80%、100%、150%、200%这个线性范围内,相对应的杂质C的浓度为0.205µg/ml、0.5125µg/ml、0.820µg/ml、1.025µg/ml、1.5375µg/ml、2.050µg/ml,在这个线性范围内其中包含了1.0µg/ml,就证明了我们选择的主成分的进样浓度是能够保证杂质能够被定性、定量检测出来的。当然我在这里只是举了一个杂质为例。如果有好几个杂质,处理起来的情况依然类似)
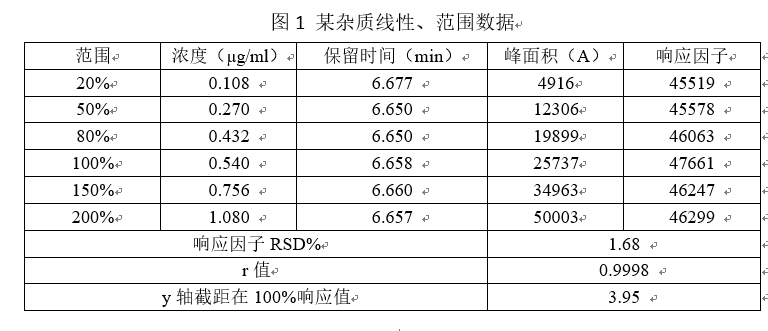
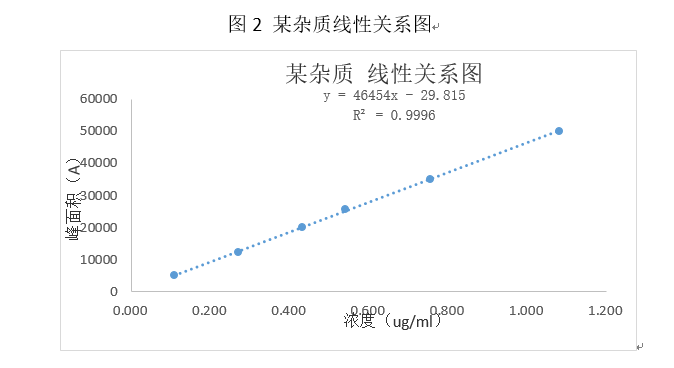
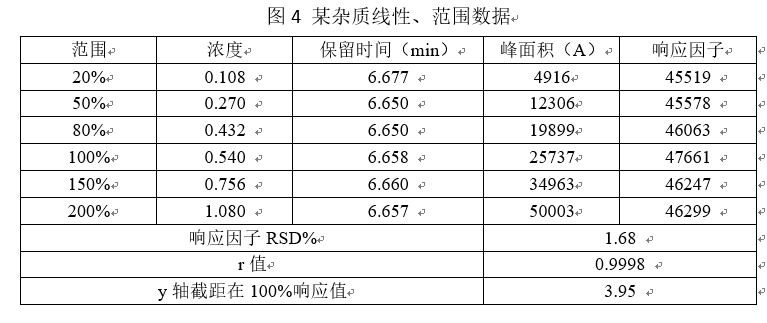
上述图表展示出,该杂质在0.108 ~1.080 µg/ml的浓度范围内呈线性相关,在该浓度范围内能准确测定出该杂质的浓度。
数据要求:应列出回归方程、相关系数、残差平方和、线性图(或其他数学模型)。r值不小于0.999,响应因子的RSD%小于10%,Y轴截距在100%处的响应值不大于2.0%(注:如果用标准曲线法,每次称量5种浓度的对照品做标准曲线,再根据标准曲线方程计算产品含量,此条不必理会。但实际情况是,我们在做线型以及范围的时候是只称取一次对照品,然后经过逐步稀释,然后制成检测主浓度20%、50%、80%、100%、150%、200%的几种溶液。然后用测得的峰面积带入曲线方程中计算浓度,该标准曲线按照原则上是过原点的,但是实际情况得到的曲线都是不过原点的,那么此时的曲线与真实的曲线就会存在误差,一般认为小于2.0%是可被接受的,但是大于2.0%那么误差就很可观了,不能被忽视)。2020版药典要求计算残差平方和,如何计算,请各位同仁查阅相关资料。
范围:分析方法的范围是样品中被分析物的较高浓度(量),和较低浓度(量)之间的一个区间,并已经证实在此区间内,该方法具有合适的准确性、精密度和线性。故线性与范围这两个实验可以一起做,以此来节约实验时间。
在计算杂质杂质含量时,到底如何选择是用自身对照法还是外标法?
举个栗子个项目中,杂质C在主成分为0.5mg/mL的进样浓度依然无法检出,整个图谱中只有主峰,此时该如何选择到底该用哪一种计算方式呢?假如杂质C的定量限为0.5μg/mL,我们设置的一系列线性的浓度为20%、50%、80%、100%、150%、200%。那么我们做方法学的时候就不能单独考虑杂质C的线性,此时还需要考察主成分的线性情况,同时我们还要考察主成分与杂质C的定量限的大小。比如当杂质C的定量限=0.5μg/mL,主成分的定量限=0.2μg/mL,那么此时表明,即使当进样浓度刚好达到主成分的定量限,杂质C依然能被定量检测出;反之,杂质C的定量限小于主成分的,那么此时如果进样浓度刚好达到主成分的定量限,杂质C则不能被定量检测出。如果杂质C与主成分的线性关系的斜率相近(具体为:杂质斜率/主成分线性的斜率=0.9~1.1之间),那么用自身对照法或者外标法均可以,但是还需要精密度实验进行考察。
具体实验操作为:在进行精密度重复性实验时(进样溶剂、与杂质定量限相同浓度的杂质对照品2针,6针主成分浓度为标准进样浓度+杂质C定量限浓度的样品,同时进自身对照6针,)用自身对照法、外标法分别计算测出杂质C的浓度。如果两者计算出的浓度相差不大于2%,那么外标法与自身对照法都可,一般为了方便,均选择自身对照法。如计算结果相差甚远,那么以外标法计算为准。(这里就不举例了,因为这种情况很少见,一般情况下,杂质的杂质C与主成分的线性关系的斜率都是非常相近的)
四、准确度
准确度:目的是为了考察实测结果与理论值之间的接近程度。(药典中做法为:原料药用已知纯度的对照品或供试品进行测定,或用所测定结果与已知准确度的另一个方法测定的结果进行比较。制剂可在处方量空白辅料中,加入已知量被测物对照品进行测定。如不能得到制剂辅料的全部组分,可向待测制剂中加人已知量的被测物进行测定,或用所建立方法的测定结果与已知准确度的另一个方法测定结果进行比较。)有关物质检测方法学验证中,准确度实验一般用回收率来表示。

当然,在做实验的时候,有可能在产品的初期,我们的样品里面是没有待测得杂质,可是随着时间的推移,可能受到温度、光照等的一些因素的影响,样品中会出现一些我们不太希望出现的物质,我们称其为有关物质,而这些物质是应该我们在实验的初期就应该预料到的一些物质,做仿制药的同事,可以参考原研药品中的一些杂质,当然也要对比仿制药的工艺路线来确定。有些杂质是一开始就存在在我们的制剂中的,而也有些杂质是后期降解产生的,那么为了验证我们的检测方法的准确性,这里就用到了加样回收,所以准确度的实验一般用回收率来表示。
有关物质回收率的具体做法:同样以上述某项目为例,上例中主成分的进样浓度为1.0mg/mL,而杂质C为一般杂质,其限定浓度定为0.1%,那么杂质C的最大进样浓度就为1.0µg/ml(因为超出这个浓度,你的样品就不合格了)。我在做有关物质回收率的时候,一般做3个水平的,那就是加入浓度的50%、100%、150%三个水平。假如样品中没有杂质C,那么做加样回收就是在样品中加入杂质C,使样品的进样时含杂质C的浓度分别为0.5µg/ml、1.0µg/ml、1.5µg/ml,50%、100%、150%三个水平已经包含绝大多数的情况了,当然如果你不放心,也可以扩大范围 例如20%、50%、80%、100%、150%、200%、300%,当然这需要你付出大量的时间与精力。不过值得注意的是,我们做实验时每个水平不能只做一次加标回收,凡事讲究要排除偶然性,所以我个人建议每个水平做3次平行实验,以排除偶然性。
图3 以杂质C为例的数据展示
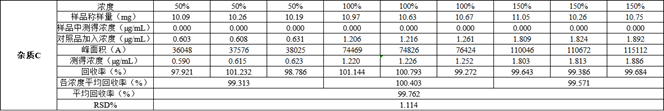
数据要求:每一次实验的回收率均应在95%~105%之间,9次实验回收率的RSD%值应小于2%。
五、精密度
分析方法的精密度值的是在规定条件下对均质样品多次取样进行一系列检测结果的接近程度(离散程度)。精密度可以从三个层次考虑:重复性、中间精度、重现性。精密度通常以多次测量结果的变异性、标准偏差或变异系数来表达。
重复性:重复性指在同样的操作条件下,在较短的时间间隔的精密度:也称为间隙测量精密度。重复性实验考查的是仪器的精密程度。
重复性论证方法:
(1)在方法规定的线性范围内至少测定9次(如3种浓度/每种浓度重复测定3次)。
(2)至少用线性100%时杂质样品测定6次。一般要求连续进样6针,以峰面积计算6针样品的RSD%要小于2.0%,一般重复性实验与线性、范围实验一起做,重复性的进样浓度为线性100%处点浓度。
个人更加推荐第二种做法,因为更加节约时间与精力。
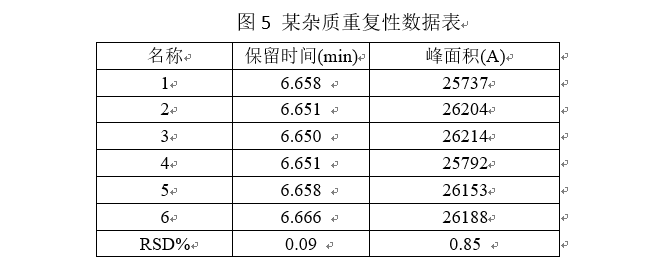
中间精密度:中间精密度是指实验室内部条件的改变,如不同日期、不同试验分析者,不同仪器等情况下测得杂质含量的精密度。一般需要更换不同的试验仪器、在不同的日期做、不同的实验人员。
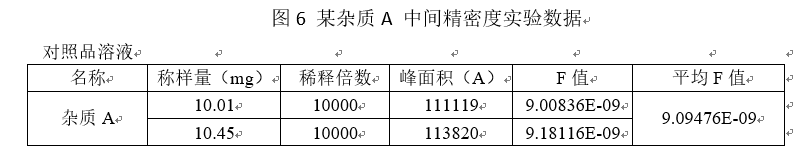
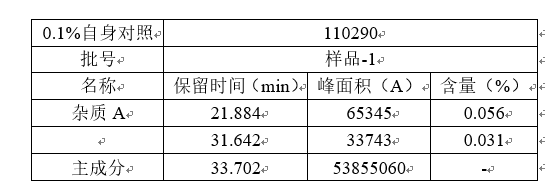
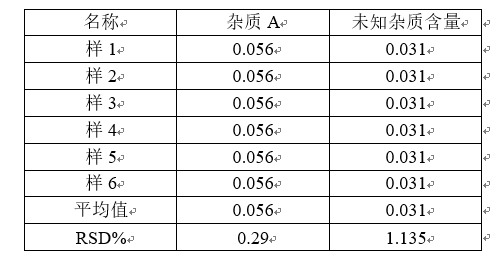
重现性:重现性是指不同实验室之间的精密度(合作研究,通常用于方法学的标准化),在做方法学的时候,是不做重现性的。
总结:做精密度只需要做重复性、中间精密度这两个实验。中间精密度的做法同重复性实验。
六、耐用性
分析方法的耐用性是指在实验参数被故意地发生细小的改变时,检测不受影响的能力,用于说明正常使用的可靠性。如果测量结果对分析条件的变化是敏感的,那么该分析条件就应该适当控制或者方法中预先注明。通过耐用性评估,建立了一系列的系统适用性参数(如分离度试验),以确保在任何使用该分析方法都是有效的。典型的变化例子如下:
——供分析用溶液的稳定性;(这个试验需要单独做,名字就叫溶液稳定性,包括含量测定的供试品溶液、对照品溶液、有关物质测定的系统适用性溶液、供试品溶液、对照溶液,都需要做相应的溶液稳定性试验,一般做到48h足矣.)
液相色谱条件下的典型变化例子:
——流动相pH值得变化:如±0.2、0.5,根据需要变动。
——流动相组分的变化:如有机相:盐相变化±2%。一般需要做3个条件,分别为正常比值、有机相+
2:盐相-2、有机相-2:盐相+2。
——不同色谱柱:(不同厂家、批号或供应商)一般做3根色谱柱的。
——不同柱温:一般温度变化为±5℃,如25℃、30℃、35℃。
——不同检测波长:一般波长变化±2nm,如273nm、275nm、271nm
——不同流速:一般流速变化为±0.1ml/min,如0.9、1.0、1.1ml/min,个人建议做这些就足够了,如果
你认为你自己的方法需要更好的耐用性,可以再扩大验证的范围。
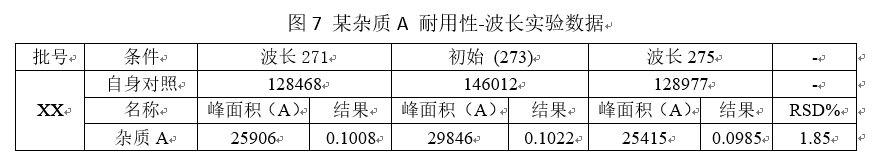
七、系统适用性试验
系统适用性试验是许多分析方法的必要组成部分。该试验是把分析设备、电子仪器、试验操作和被分析样品作为一个整体来进行评估。一般在做有关物质测得的时候需要做系统适用性试验,而且是必须要做的,目的是为了考查,仪器、色谱条件、环境等的组合能否在此条件下,将样品中的有关物资分开,系统适用性实验。一般含量测定无需做系统适用性试验。
在做方法学验证的时候,个人认为我们的系统适用性溶液需要连续进样6针,统计各种杂质以及主成分的保留时间、峰面积,同时计算保留时间、峰面积的RSD%值,来确定你的仪器、检测方法的稳定性。当然在同一时间段、同一实验情况下你也需要单独进样系统适用性溶液中的每一种杂质与主成分,进样浓度要与系统适用性溶液每一种杂质的浓度相同或者大致相同,记录保留时间、峰面积。通过计算各种杂质保留时间、峰面积的RSD%值,去证明你的检测方法、检测仪器是适用的。
总结:上面写的内容只是我一家之言,具体的品种需要做的方法学也不尽相同,大家只要把握主主要方向——用你的数据去说服CDE的老师,实验的设计千千万万,只要你能说明你的检测方法是适用的,条条大路都是通罗马的,所以做实验不要亦步亦趋,也不要固步自封,多多学习、多多的思考才能紧跟时代潮流,最后祝愿打击实验顺利。
<END>


















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论