































《手性药物质量控制研究技术指导原则》指出,因为立体异构体的数量与手性中心的数目成指数关系,在手性中心较多(一般大于或等于3)时,仅通过终产品的质量标准来控制所有的立体异构体杂质,在技术上有一定的难度,有时甚至做不到。这时一定要根据工艺中手性中心的引入方式,合并采用源头控制及生产的过程控制,来全面控制产品的光学纯度。这样既能有效地控制产品的光学纯度,又能合理地降低终产品质控的难度。
上面指南中的理论指导,虽然看懂了,但如何实操,仍然迷茫。本人目前已申报了多个多手性中心药物,且多个品种已经获批上市。以下选取了比较典型的1个品种,该品种含有5个手性中心,该品种的手性中心均为多个起始物料引用,且工艺过程中手性中心不参与反应。
案例的思路
虽然API中手性中心比较多,但通过工艺路线溯源,其源头手性化合物的手性中心个数骤减。最简单的方式为采用源头控制,但绝大部分起始物料的产品质量很难将所有的异构体杂质限度做到鉴定限以下(常规0.10%以下),故成品中也需考虑进行异构体的研究。粗暴的方式为通过终产品的质量标准来控制所有的立体异构体杂质,但在技术上难度极大,甚至做不到。采用源头控制,并通过成品中概率推算的方法,计算风险较大的异构体杂质,再针对较大风险的杂质,在成品中建立方法进行研究。
实例
通过对源头(起始物料)控制及过程分析,以满足在成品中控制的目的。具体研究如下:
某药物API中含有5个手性,均由API合成的起始物料引入,其中起始物料SM1引入3个手性中心,起始物料SM2引入2个手性中心。这2个起始物料中存在的对映异构体及非对映异构体可能传递至API中。
(1)源头(起始物料)控制
起始物料SM1含有3个手性中心,理论上存在1个对映异构体和6个非对映异构体(3对)。起始物料SM1中对3对非对映异构体和对映异构体杂质均作为已知杂质进行了控制,限度均不得过0.10%。
起始物料SM2含有2个手性中心,理论上存在1个对映异构体和1对非对映异构体,起始物料SM2中1个对映异构体和1对非对映异构体均作为已知杂质进行了控制,限度分别为不得过0.15%和0.10%。
(2)过程分析
起始物料SM2中在对映异构体项下控制SM2-01不得过0.15%, SM2-03(一对)在有关物质项下控制不得过0.10%。
SM1含有3个手性中心,理论上存在1个对映异构体SM1-08和6个非对映异构体SM1-05(一对)、SM1-06(一对)、SM1-07(一对)。SM1-08在对映异构体项下控制不得过0.10%,SM1-05(一对)、SM1-06(一对)、SM1-07(一对)在有关物质项下控制均不得过0.10%。
结合工艺路线可知,手性中心不参与反应,无强酸强碱剧烈反应条件,不存在手性中心翻转的条件。故某药物理论上可得25共32个化合物,其中1个为对映异构体杂质,30个非对映异构体杂质。
假设起始物料及其异构体杂质100%参与后续反应,且无手性翻转的前提下,在某药物成品中可能生成的异构体杂质,理论含量计算结果如下:
- 100%(SM1)×0.15%(SM2-01)=0.15%(1个非对映异构体)
- 100%(SM1)×0.10%(SM2-03)=0.10%(1对对映异构体)
- 0.10%(SM1-08)×100%(SM2)=0.10%(1个非对映异构体)
- 0.10%(SM1-08)×0.15%(SM2-01)=0.00015%(1个对映异构体)
- 0.10%(SM1-08)×0.10%(SM2-03)=0.00010%(1对对映异构体)
- 0.10%(SM1-05)×100%(SM2)=0.10%(1对对映异构体)
- 0.10%(SM1-05)×0.15%(SM2-01)=0.00015%(1对对映异构体)
- 0.10%(SM1-05)×0.10%(SM2-03)=0.00010%(2对对映异构体)
- 0.10%(SM1-06)×100%(SM2)=0.10%(1对对映异构体)
- 0.10%(SM1-06)×0.15%(SM2-01)=0.00015%(1对对映异构体)
- 0.10%(SM1-06)×0.10%(SM2-03)=0.00010%(2对对映异构体)
- 0.10%(SM1-07)×100%(SM2)=0.10%(1对对映异构体)
- 0.10%(SM1-07)×0.15%(SM2-01)=0.00015%(1对对映异构体)
- 0.10%(SM1-07)×0.10%(SM2-03)=0.00010%(2对对映异构体)
从上述的理论计算所得的31个杂质含量可知,其中1个为对映异构体杂质的理论含量为0.00015%,30个非对映异构体杂质中只有1个杂质含量为0.15%,9个含量为0.10%,其他均为0.00015%或0.00010%。
对于理论杂质最大含量为0.15%的非对映异构体杂质(代号为杂质G),我公司在起始物料SM2中进行了加标试验,在起始物料SM2中加标杂质SM2-01,按照确定的生产工序进行后续反应,试验数据如下:
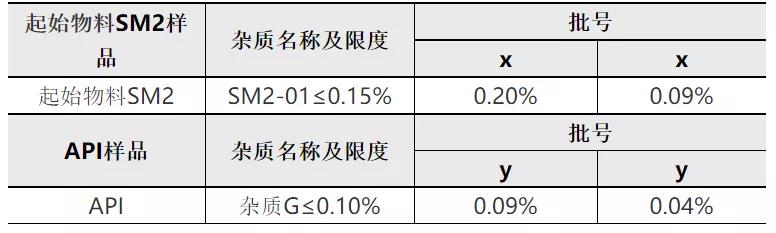
从上表可知,当起始物料SM2中SM2-01含量为0.09~0.20%%时,按照既定生产工艺进行生产,制备所得API中杂质G为0.04%~0.09%。上述结果说明后续步骤对SM2-01杂质及传递杂质具有一定的消除能力。结合本品工艺对该杂质的消除能力,在起始物料SM2中控制该杂质≤0.15%,可确保成品中相应异构体含量低于0.10%。
结合手性中心构型反转的机理来考虑,当手性中心上存在一个活性氢原子,且邻位有羰基等吸电子基团时,在酸、碱或光照等条件下,该手性中心就可以通过分子内的互变异构而发生外消旋化。结合本品的工艺条件及起始物料、中间体及成品的结构可知,各化合物上的手性碳原子均不会参与反应,且本品制备工艺条件均较温和,无强光照射、强酸和强碱等剧烈的反应条件,故各手性中心不存在翻转生成异构体的可能。另通过起始物料SM2中杂质SM2-01传递反应生成杂质G,证实了在工艺研究过程中,无手性中心发生翻转导致异构体杂质增加的情况。
结合某药物的工艺路线可知,手性碳原子均未参与后续反应,SM1和SM2的手性结构能够在某药物的分子结构中得到保持。
故通过对起始物料SM2和SM1中异构体杂质的控制,达到了对某药物中的异构体杂质控制的目的,已定控制策略可确保异构体杂质对成品质量无风险。
(3)异构体杂质的稳定性研究
本品引入的异构体杂质除手性中心的立体构型外,其平面结构均与API相同。本品API的有关物质方法为高效液相色谱法,采用紫外检测器。在紫外吸收中,平面结构相同其紫外吸收一致,故本品的有关物质分析方法对异构体杂质均具有检出能力。
结合有关物质分析方法对异构体杂质的检出能力,通过长期24个月及加速6个月稳定性考察期间未知杂质均未出现增长趋势,故可推断样品在既定的贮藏条件下未出现手性中心构型翻转。
另结合手性中心构型反转的机理来考虑,当某手性中心上存在一个活性氢原子,且邻位有羰基等吸电子基团时,在酸、碱或光照等条件下,该手性中心就可以通过分子内的互变异构而发生外消旋化。某药物的结构中不存在活性氢原子,且邻位有羰基等吸电子基团,另本品包装和储存条件为“药用低密度聚乙烯袋内包装,尼龙扎带封口,复合膜袋热封,纸板桶外包装保存;遮光,密封保存”,本品的储存条件能够确保API在储存条件下不会被酸、碱或光照等条件破坏。故稳定性放置过程中异构体杂质不可能产生。
为了进一步评估API非对映异构体是否有增加的风险,在加速稳定性试验过程中对存在风险最大的非对映异构体杂质G的含量进行研究,检测数据见下表:
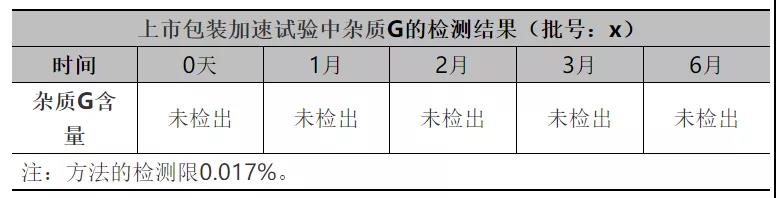
从上表数据可知:稳定性过程中杂质G含量无增加趋势,进一步说明稳定性过程中异构体杂质不会通过翻转降解产生。本品稳定性过程中异构体杂质无降解增加的风险。
综上,成品中各异构体杂质已得到合理控制,对成品质量无影响。
上述案例为本人申报上市的实际品种,且已获批,上述的研究思路是被官方认可的。当初申报阶段,和其他公司聊该品种,几个批准的公司都是在成品中研究了多个异构体。本人当时万分担忧异构体研究发补。方法真是太难了,分析部门做不到啊。
另针对手性中心参与反应的品种,需要考虑该案例的思路进行分析及生产的过程控制,二者结合进行分析。
上述研究思路可能还存在瑕疵,整体个人认为是科学合理的,供同行们参考。
<END>



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论