































目前,《中国药典》2020年版四部通则增修订内容已经公示了十三批,对通则中的多数内容进行了修订,在第三批增修订内容(2018-12-28)中公布了9101分析方法验证指导原则(第一次征求意见稿)。作者对这次征求意见稿中的检验项目和验证指标以及正文中的一些规定进行了简要分析,供研发同仁参考,但本文中的一些观点不能代表官方意见,需谨慎使用。

一、专属性
专属性系指在其他成分(如杂质、辅料等)可能存在下,采用的分析方法能正确测定出被测物的能力。专属性是大多数分析方法中最重要的指标,是建立分析方法首先要考虑的问题。色谱法中常见的专属性考察指标为分离度。
在分析方法验证过程中,有很多方法来考察专属性:
(1)分离度考察(各起始物料、中间体、副产物、降解产物、主成分、空白辅料、溶剂),对于有关物质而言,已知杂质或降解产物应加入到新配制的含活性成分的溶液中,确保与被测定峰完全分离,主峰与相邻杂质,以及杂质与杂质之间的分离度应大于1.5(方法开发时可以规定大于2),有时在标准中规定采用相对保留时间的方法确定某个杂质,那么在专属性考察中应统计相对保留时间,并注意在其他验证项目中也进行统计。
(2)降解试验(光照、高温、高湿、酸碱水解、氧化)或加速试验中的样品。
(3)采用PDA(或DAD)检测器、质谱检测器(MS)进行峰纯度检查。
(4)改变方法中的某个条件进行验证(如在等度洗脱后增加有机相比例,考察有无未洗脱下来的新杂质;采用CAD检测器等考察无UV吸收的其他杂质峰)。
(5)考察未精制的粗品,并与最终精制过的成品进行杂质谱对比,根据粗品中各成分的分离度和样品中可能出现杂质来优化色谱参数。
(6)代表性制剂样品(制剂工艺中可能新增其他杂质)。
上述方法中,值得讨论的是降解试验。指导原则中没有关于降解条件的特别规定,因此做法各不相同。通常,原料药的强制降解试验包括液态的和固态的情况(应包括酸/碱水解,高温,高湿,氧化和光照),仅通过原料药的溶液或固体的降解一般无法准确推断制剂中药物的降解情况,因为制剂中的非活性成分(辅料)也可能与原料药反应或催化降解反应。对于制剂,常用试验条件为高温、高湿和光照。制剂的降解很大程度决定于该制剂是溶液还是固体。对于固体制剂,主要试验是高温、高湿和光照降解,最常见的反应类型是水和原料药的反应,因此高温和高湿组合试验也很关键。制剂降解中需同时进行空白辅料的降解试验。
降解程度通常是使主成分有5%~20%的降解(主峰剩余80%~95%),尽量不要让降解超过20%,如果超过20%,则应改变降解强度和/或时间。避免使用高浓度或剧烈的破坏条件,尽量使用低浓度、长时间的条件进行降解。因为当使用剧烈条件时可能出现次级降解,药物的降解机理可能会发生改变。
酸降解:酸降解可以通过在样品溶液中或固体(原料药或制剂)中加入0.1mol/L盐酸溶液进行,条件可以在室温或60℃条件下最长放置3天。
碱降解:碱降解可以按照酸降解的方式进行,如0.1mol/L氢氧化钠,二者均应采用相应的酸碱溶液进行中和。
氧化降解:亲核氧化反应可以在样品溶液中或固体(原料药或制剂)中加入稀过氧化氢溶液(如3%),在室温下反应24h进行,可能产生N-氧化物或亚砜杂质。自由基氧化可以使用偶氮二异丁腈(Azobisisobutyronitrile,AIBN)来进行研究,在40℃下反应1-3天。应根据药物的氧化降解机理选择适当的氧化剂。
高温降解:高温降解试验可以采用样品溶液中或固体(原料药或制剂)在高温下进行,典型条件是在60°C 进行24-48 h, 基于贮藏条件,其他室温以上的条件也可以采用。
光照降解:高温降解试验可以采用样品溶液中或固体(原料药或制剂),试验条件可根据ICHQ1B中的光源应包括可见光和紫外光。
如果化合物难溶于水,可加入其他助溶剂使其在盐酸或氢氧化钠溶液中或过氧化氢溶液中溶解。助溶剂的选择应基于药物的结构。两种最常用的共溶剂为乙腈和甲醇。但应当注意的是,由于受诸多因素的影响,通过助溶剂增加溶解度并非都能加速降解。选择合适的共溶剂时应特别关注药物的化学结构。应能够识别药物结构中易反应的化学位点,切勿选择能与药物发生反应的共溶剂。例如,当药物结构中含有羧酸、酯键、酰胺键、芳香胺或羟基时,应避免使用甲醇和其他醇类作为共溶剂进行酸降解试验。这样会避免因药物与甲醇或其他醇类反应导致的不必要的研究工作。另外,只有当乙腈比例为20%或更低时才可与1N NaOH溶液混合,NaOH浓度降低至0.1N时可以使用更高比例的乙腈作为共溶剂。乙腈是最常用的共溶剂。
对于制剂而言,高温、光、湿(RH70%或80%)、高温+高湿是很重要的,因为这些条件更能够模拟今后制剂中遇到的实际情况,发现这些条件下产生的杂质可以提前做出是否需要增加这些杂质研究的判断。例如,作者曾在高温高湿条件下发现有三个新增杂质,对过期的参比制剂以及经过加速试验的参比制剂进行检测时发现,产生的杂质与高温高湿条件一致。
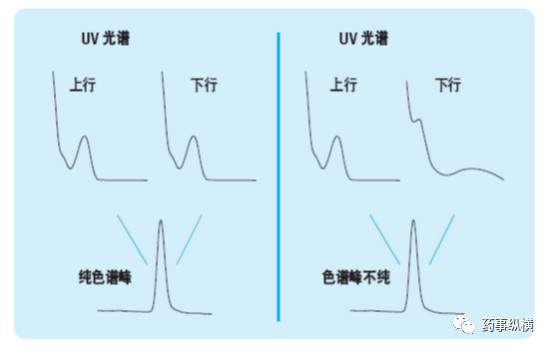
目标化合物色谱峰的峰纯度是专属性的一个重要考察指标,使用光谱检测器进行专属性评价的原理见下图,以色谱法和紫外-可见二极管阵列检测器为例。图中显示了一个专属性色谱方法和一个非专属性色谱方法。两个色谱峰看起来非常相似。从峰形来看,无法明显看出该色谱峰中包含一个或几个化合物。这两个例子中,分别在色谱峰的上行和下行处查看其UV 光谱图,归一化后进行比较。左例中,光谱图是相同的,表明该色谱峰中只有一个化合物,或者说该色谱峰是光谱纯的。右例中,两个UV 光谱图不同,由此证明该色谱峰显然不纯。现在二极管阵列检测器能够对光谱图进行自动比较,进行峰纯度评价。
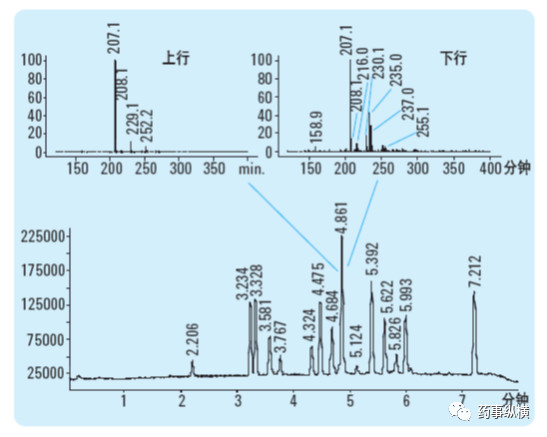
当共洗脱化合物的光谱图非常接近时,紫外-可见二极管阵列检测器就有其局限性,这种情况下,最好为HPLC 配一个质谱检测器。质谱通常比紫外光谱专属性更强,因此常用于色谱的选择性评价。一个HPLC 与质谱配合使用的例子见下图。对于4.86 分钟洗脱的色谱峰,在不同部位采集的质谱图不同,显示该色谱峰不纯。
如果文献报道药物在各条件下都比较稳定,并且在所设计的剧烈条件下降解仍达不到5%或10%,则没有必要再继续进行降解。总之,降解试验不应为了降解而降解,最好能够模拟制剂加速条件或以后可能遇到的剧烈条件,即具有一定的稳定性指示作用是最好的。如某药物对光敏感,那么在进行降解试验是应考察固体和溶液哪个更敏感、对什么光敏感、采用的避光措施能否有效避光等。
在经过定位研究及降解试验研究后,一旦证明方法的专属性符合要求,那么此时方法中的色谱柱、流动相组成、流速、柱温、检测波长就可以基本确定下来,以后可以在此基础上继续进行摸索和优化。值得注意的是,有时色谱图中显示相邻两个色谱峰的分离度为1.5,虽然数据上符合要求,但放大观察后可能会发现二者并不能完全分离,此时最好继续优化条件使杂质的分离度大于2,以免后期出现问题。
二、准确度
准确度系指所建立方法测定的结果与真实值或参比值接近的程度,一般用回收率(%)表示。回收率应在规定的线性范围内试验。
在本指导原则中,准确度的数据中有一定变化,例如将同一浓度(相当于100%浓度水平)6份供试品的方法删掉了,只保留了3种不同浓度,每种浓度分别制备3份溶液,共9份样品的测定方法。

对于化学药,将配制方法中的推荐比例也删掉了。

样品中待测成分含量和回收率限度关系可参考下表,在基质复杂、组分含量低于0.01%及多成分等分析中,回收率限度可适当放宽。
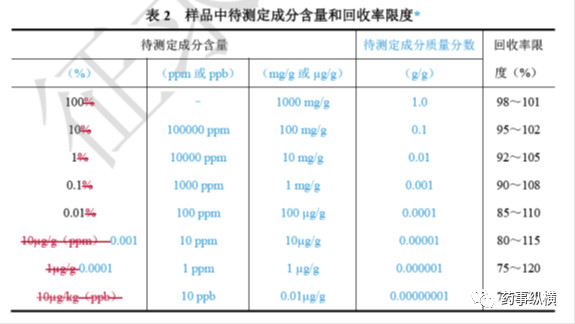
在方法学验证中,对于原料药的含量测定回收率而言,应采用已知含量(真值)的对照品配制三种不同浓度,每个浓度三份,共九份进行测定,九份结果的平均值作为最终结果(测定值),并将其与其他方法测得的结果或与对照品报告单上的结果进行比较,如果基本一致,说明方法准确性很高。对于原料药中的杂质而言,可在原料药中加入50%限度、100%限度、150%限度、200%限度的杂质对照品,每个浓度配制三份,进行评价。
对于制剂含量测定而言,可以在处方量空白辅料中加入不同量的对照品,例如正常分析中被测物含量的80%、100%、120%,对于溶出度、含量均匀度、杂质测定而言,可以在空白辅料或样品中加入一定比例的对照品或杂质对照品,浓度范围包括药典中推荐的范围,对于杂质而言一般为50%限度、100%限度、150%限度、200%限度,根据测定总量、加入量和样品中原有量,计算回收率百分比。
杂质回收率的计算时,如果最终标准是加校正因子的主成分自身对照法,应采用确定的方法计算回收率。同时,可以验证外标法和加校正因子的主成分自身对照法两种方法的测定结果是否一致,并将这两种方法的计算结果均列入方法学验证资料中。
如不能得到制剂辅料的全部组分,可向待测制剂中加入已知量的被测物进行测定。用实测值与供试品中含有量之差,除以加入对照品量计算回收率。但应注意,在加样回收率试验中须注意对照品的加入量与供试品中被测成分含有量之和必须在标准曲线线性范围之内,加入的对照品量要适当,过小则引起较大的相对误差,过大则干扰成分相对减少,真实性差。
在测定杂质回收率时,通常是配制一份杂质母液,然后在供试品中按设计的百分比浓度加入,如果杂质的线性与回收率先后在这台仪器上进行,那么可将线性试验中杂质的峰面积与回收率试验中杂质的峰面积进行对比,直观考察基质效应(辅料影响)、以及溶液制备过程对准确度的影响,如有影响,应分析原因,提高回收率。
三、精密度
精密度系指在规定的条件下,同一份均匀供试品,经多次取样测定所得结果之间的接近程度。一般用偏差、标准偏差或相对标准偏差表示。在相同条件下,由同一个分析人员测定所得结果的精密度称为重复性;在同一个实验室,不同时间由不同分析人员用不同设备测定结果之间的精密度,称为中间精密度;在不同实验室由不同分析人员测定结果之间的精密度,称为重现性。含量测定和杂质的定量测定应考察方法的精密度。
重复性试验具体做法为:在规定范围内,取同一浓度(分析方法拟定的样品测定浓度,相当于100%浓度水平)的供试品,用至少测定6份的结果进行评价;或设计3种不同浓度,每种浓度分别制备3份供试品溶液进行测定,用9份样品的测定结果进行评价。
对含量测定而言,通常重复性按照第一种方法配制6份相同浓度的供试品溶液,由一个分析人员在尽可能相同的条件下进行测试。中间精密度是由不同人员,在不同时间按照重复性的方法采用同一批样品配制6份相同浓度的供试品溶液,最后统计两次共12份含量数据的相对标准差。重复性和中间精密度应符合下表规定。
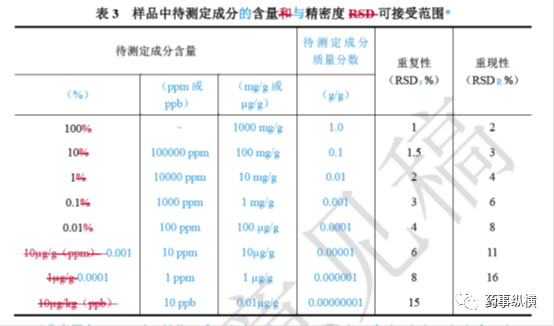
对于有关物质的重复性和中间精密度考察,除了应统计上表中规定的RSD外,也应考虑各样品的杂质数量、杂质总量,有时杂质检出很小时会统计杂质的极差,如果极差小于0.02%(或限度的20%)也可以认为无明显差异,说明方法精密度良好。
重现性是考察不同实验室间的精密度,可以在方法转移中进行评估。
值得注意的是,较高的精密度往往受到玻璃仪器、转移、称量、稀释、过滤等因素的制约,因此过程中应特别注意规范操作。
四、检测限和定量限
检测限系指试样中被测物能被检测出的最低量。检测限没有定量意义。定量限系指试样中被测物能被定量测定的最低量,其测定结果应符合准确度和精密度要求。对于杂质测定应确定方法的定量限。
指导原则中规定了三种方法测定检测限和定量限:
(1)直观法:用已知浓度的被测物,试验出能被可靠地检出(或定量测定)的最低浓度或量。
(2)信噪比法:用于能显示基线噪声的分析方法,即将已知低浓度试样检出的信号与空白样品测出的信号进行比较,计算出能被可靠地检出(或定量)的被测物质最低浓度或量。一般以信噪比3:1或2:1时(或10:1)相应浓度或注入仪器的量确定检测限(或定量限)。
(3)基于响应值标准偏差和标准曲线斜率法。按照LOD=3.3δ/S或LOQ=10δ/S公式计算,式中LOD为检测限,LOQ为定量限,δ为响应值的偏差,S为标准曲线的斜率。其中δ可以通过以下方法获得:①测定空白值的标准偏差;②采用标准曲线的剩余标准偏差或是截距的标准偏差来代替。
方法学验证中,应在申报资料中说明是采用上述何种方法来测定检测限或定量限的。
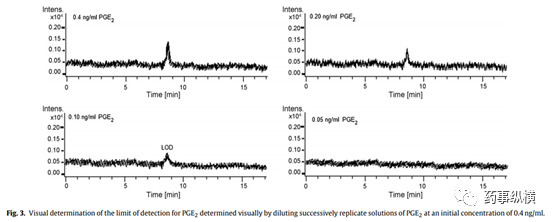
对于目测法而言,需要提供逐级稀释的图谱,并说明稀释过程。如下图采用采用初始浓度0.4ng/ml逐级稀释,最后目测观察0.10ng/ml的浓度相当于检测限浓度。
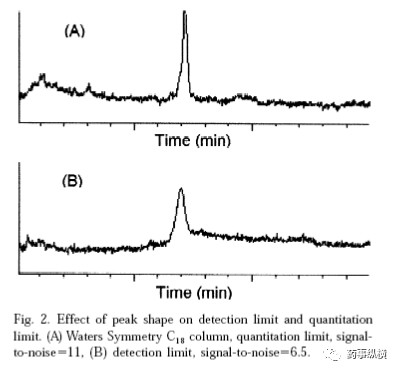
检测限和定量限常采用信噪比法测定。该计算方法下的检测限与定量限是通过对照品逐级稀释的方法获得的,没有考虑任何样品制备步骤的影响,因此其值往往比分析方法本身的实际检测限与定量限低,而且每台仪器也不一样,因此信噪比法是用来考察仪器本身性能的,与方法本身的检测限或定量限还是不一样的。信噪比法受到的影响因素比较多,例如所用色谱柱的柱效、检测器的氘灯寿命等。例如,下图中峰形尖锐的信噪比高,s/n=11,此时检测限和定量限都较低。
尽管信噪比法有诸多问题,但目前仍是方法学验证中最常用的计算方法。有时,为了保证方法的灵敏度,有时可采用灵敏度较差的PDA/DAD检测器进行检测限和定量限的考察。
通常检测限应低于限度的1/30,定量限应低于限度的1/10,如果达不到这一要求,可适当放宽,只要规定定量限应比ICH规定的杂质的报告限低即可(例如50%报告限)。
由于灵敏度受仪器、色谱柱、试剂、泵系统、检测器等的影响很大,因此为了保证方法的检测能力,有些药物的质量标准中规定了灵敏度溶液的信噪比作为系统适用性要求来考察整个色谱系统的灵敏度,相信这种方法以后会应用越来越多。
在指导原则中还有一句话很重要:“上述方法获得的检测限和定量限数据须用含量相近的样品进行验证,应附测试图谱,说明测试过程和定量限结果,包括准确度和精密度验证数据。”如何进行检测限和定量限的验证呢?常见的做法是采用杂质加标的方法进行验证,例如在供试品溶液中加入相当于检测限或定量限浓度的杂质,考察加入后的色谱图中信噪比是否符合要求。定量限的精密度验证目前基本都做到了,常采用配制6份定量限浓度的溶液,所测6份溶液杂质峰保留时间的相对标准差应不大于2.0%,峰面积的相对标准差应不大于5.0%的方法进行评价。有时也进行定量限浓度的准确度验证,可接受限度可参考上述准确度项下的规定,有时也可以适当放宽。
对于滴定方法而言,不必验证方法的检测限和定量限,而应更重视方法的准确性、精密度和专属性。
五、线性
线性系指在设计的范围内,测定结果与试样中被测物浓度直接呈比例关系的程度。在9101分析方法验证指导原则中规定了两种测定方法:
(1)同一对照品溶液稀释
(2)分别配制对照品溶液
并规定至少采用5种不同浓度进行试验,以响应信号作为与被测物浓度的函数作图,首先观察是否呈线性,再用最小二乘法进行线性回归。用于结果计算的线性回归方程其截距不得明显偏离零点。如果得到一个显著的非零截距,应证明其对方法准确度没有影响。对于化学药的含量测定,线性试验可接受范围为:回归线的相关系数(R)不得小于0.998,Y轴截距应在100%响应值的2%以内,响应因子的相对标准差应不大于2.0%。对于有关物质测定,线性试验可接受范围为:回归线的相关系数(R)不得小于0.990,Y轴截距应在100%响应值的25%以内,响应因子的相对标准差应不大于10%。
方法(1)采用同一对照品溶液(贮备液)进行系列稀释,与分别配制几种浓度的对照品分别进样相比能够减少误差,因此是做线性的首选方法。
在进行杂质研究中,经常要测定校正因子校正因子,目前首选方法是标准曲线法:精密称取杂质对照品和主成分对照品,分别制备系列溶液(涵盖定量限、标准限度),分别进样后,按最小二乘法以进样量对响应值(峰面积等)进行线性回归,求得两条标准曲线,两曲线斜率之比即为校正因子。由于从定量限到标准限度往往超过一个或多个数量级,此时通过最小二乘法存在一定的缺陷,高浓度对结果的影响很大,分段进行计算时,往往低浓度不呈线性。因此,此时药典中规定的首先观察就很有意义,如果明显不呈线性,计算出的结果也是不对的。除了观察之外,也可以采用低浓度点的峰面积对浓度计算单浓度点的校正因子,如果真的呈线性,那么每个点的校正因子应基本一致,如果差异很大说明是“假线性”。上述内容可以用下图来比较直观的说明。
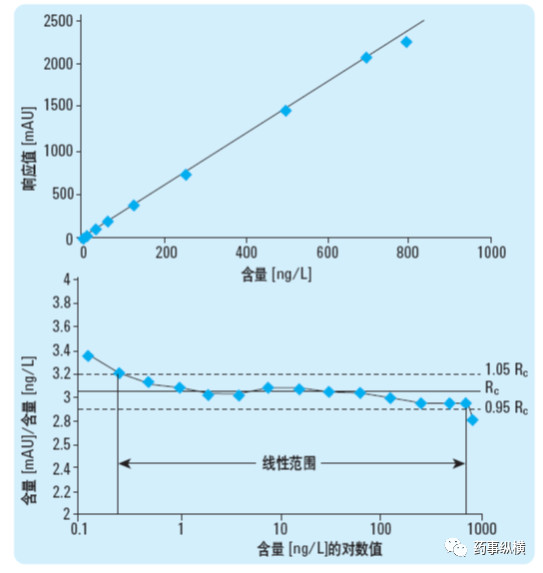
校正因子的验证试验很重要,在测定时应至少采用上述线性方法重复测定三次校正因子,比较三次结果的一致性。如参考了药典标准条件,应将实测的校正因子与药典质量标准中的校正因子进行对比。
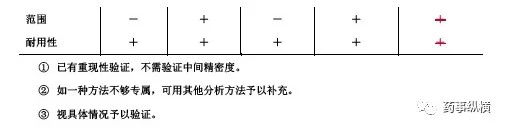
六、范围
范围系指分析方法能达到精密度、准确度和线性要求时高低限浓度或量的区间。范围应根据分析方法的具体应用及其线性、准确度、精密度结果和要求确定。
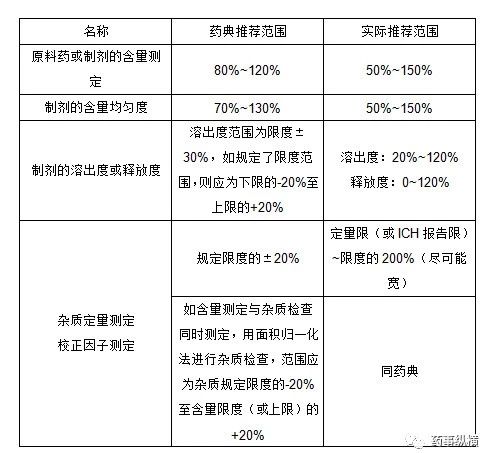
范围推荐值
七、耐用性
耐用性系指在测定条件有小的变动时,测定结果不受影响的承受程度,为所建立的方法用于常规检验提供依据。
耐用性通常对应ICHQ2指导原则中的Robustness,这种改变是微小的、故意对方法本身参数进行的改变,考察对结果的影响,在方法开发阶段、预验证阶段就应进行考察。如不同流动相组成、不同品牌色谱柱、不同检测波长、不同pH、不同柱温、不同流速、不同进样口和检测器温度等。常见的改变包括:考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、检测波长变化±2nm、流速相对值变化±20%以及采用三根不同品牌的色谱柱进行测定,考察系统适用性和样品测定结果的变化。
此外,耐用性还应包括Ruggedness,即方法在实际应用条件下结果的重现性。这些影响因素包括不同分析人员、不同实验室、相同厂家相同品牌不同批号色谱柱、新色谱柱、不同品牌仪器、不同厂家的化学试剂等。这些变量在以后方法使用中更为重要,因此也应该在方法开发和验证阶段就重视。在方法转移阶段,方法的ruggedness应该特别关注,并与同批样品之前的结果进行对比。在后期方法使用的过程中,ruggedness会经历大量的检验来评价,应注意问题汇总。例如,如果发现相同品牌不同批号的色谱柱有影响,就应该保存好这些“好”的色谱柱,多从厂家采购相同批号的色谱柱,以避免以后可能出现的问题。
最后,溶液的稳定性、样品前处理方法、流动相的稳定性等也应进行考察。通常对照品溶液、供试品溶液都要考察溶液稳定性,一般而言,24小时内的溶液稳定性是需要考察的。对于室温下不稳定的,可以选择冰箱内保存(2-8℃)。对于含THF的流动相容易被氧化,应现配现用。某些磷酸盐或醋酸盐在夏天时会因为微生物生长,因此应不宜使用过长。
八、系统适用性试验
《中国药典》2020年版编制大纲要求“加强方法中系统适用性试验研究并在标准中予以体现,提高方法的重现性和准确性”,但在本通则征求意见稿中没有明确规定需要进行系统适用性试验研究和验证,仅在耐用性项下规定:测定条件小的变动应能满足系统适用性试验要求,以确保方法的可靠性。目前,在方法开发与验证的过程中,存在着对系统适用性试验认识和研究不足、缺少重视的情况,应引起重视。
参考文献
[1] 9101分析方法验证指导原则(第一次征求意见稿)
[2] 安捷伦.分析方法验证基础导论
[3] 张哲峰.HPLC法校正因子研究中的几个问题. CDE电子刊物,20111207
[4] 黄晓龙.有关物质分析方法验证的可接受标准简介.CDE电子刊物,20060208
[5] 黄晓龙.含量测定分析方法验证的可接受标准简介.CDE电子刊物,20060120
[6] 王思寰,吴越,王玉,靳桂民,洪小栩.药品分析方法验证药典的讨论.药物分析杂质,2018,38(9)1646-1650
[7] Pedro Araujo.Key aspects of analytical method validation and linearity evaluation. Journal of Chromatography B, Volume 877, Issue 23, 1 August 2009, Pages 2224-2234
[8] 张哲峰,成海平,宁黎丽,田洁.CTD申报资料中杂质研究的几个问题.CDE电子刊物,20121226
[9] L.R森德尔等.实用高效液相色谱法的建立(第二版).华文出版社



















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论