
































近年来由于核酸修饰和递送载体的突破,带来了变革性疗法的创新浪潮,其中被认为是继小分子药物、抗体药物之后第三代创新药物的核酸药物迎来了爆发式增长,其优势在于广泛的可成药靶点、特异性强、安全性高、效果持久、开发成功率高和制造成本低等。寡核酸药物是由20-60个核苷酸单元组成的单链或双链结构,通过作用于mRNA来调控疾病基因转录翻译,抑制基因表达,主要有反义寡核苷酸 (ASOs),小干扰RNA(siRNA)和微小RNA(miRNA)。
一、寡核苷酸的固相合成
目前寡核酸药物大多通过固相亚磷酰胺化学法进行合成。化学合成按照3'-5'的方向进行。
常用的固相载体为可控微孔玻璃珠(CPG),CPG通过linker与初始核苷酸核糖的3'-OH共价结合,而核糖的2'-OH用诸如叔丁基二甲基硅基(TBDMS)的保护试剂进行保护,5'-OH则用双甲氧基三苯甲基(DMT)保护。此外,由于腺嘌呤、鸟嘌呤和胞嘧啶存在伯氨基团,也需要用酰基试剂(例如苯甲酰基)进行保护。
固相合成每个循环主要包括四个步骤:去保护、偶联、氧化和加帽。合成中产生的杂质(包括最后的脱保护)有以下几种类型:
- 截短的序列(shortmer sequences)N-X,如N-1,N-2;
- 更长的序列(longmer sequences)N+X,如N+G,N+A;
- 硫代磷酸酯键(PS)产生的非对映异构体和氧化产生PO杂质;
- 胞嘧啶和5-甲基胞嘧啶脱氨基引起的杂质;
- 其他杂质,如碱基脱嘌呤杂质、2'-5'连接异构杂质、序列异构体等;
下文对上述杂质的产生及其分析和产品质量控制做简单介绍。
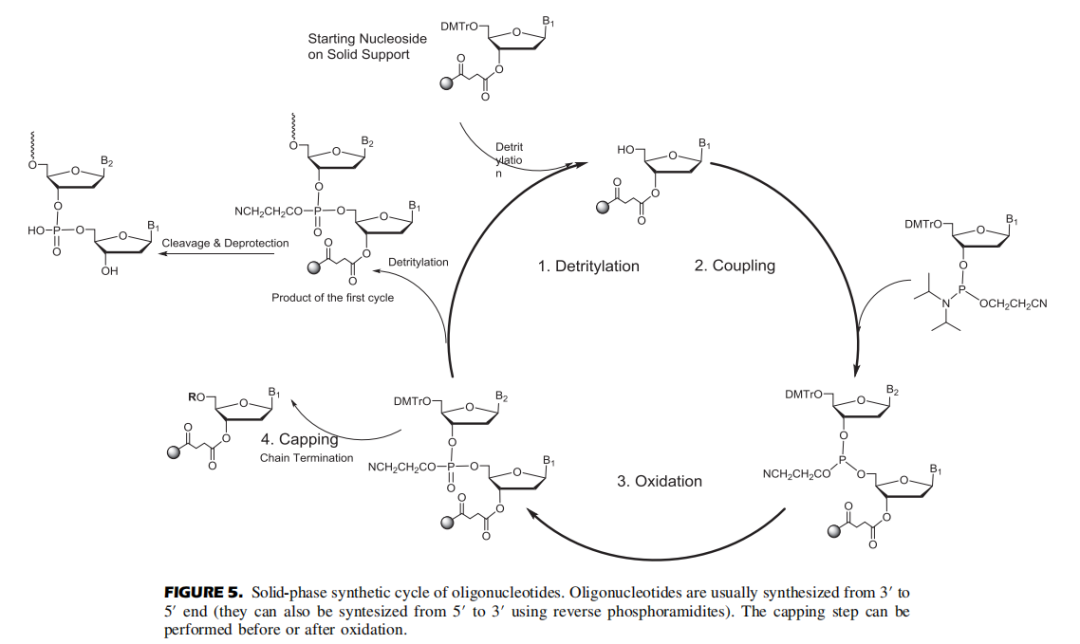
▲寡核苷酸的固相合成
01.脱三苯甲基(Detritylation)
使用溶解在二氯甲烷/甲苯中的2-3%三氯乙酸(TCA)或二氯乙酸(DCA)移除核糖5'的DMT基团,暴露5'-OH,以供下一步偶联。脱保护时间取决于流速和柱子尺寸,反应时间不够/脱保护剂酸性太弱会产生N-1杂质(与完整长度为N的寡核苷酸相比仅相差一个核苷酸);反应时间太长/脱保护剂酸性太强则导致RNA中脱嘌呤的产生。反应完成后,用乙腈洗涤去除残留的脱保护剂,此步骤中乙腈含水量一般小于20 ppm,脱保护试剂冲洗不干净导致N+1杂质的产生。值得注意的是,DCA溶液中的痕量三氯乙醛杂质与合成中的寡核苷酸末端游离羟基反应导致比全长产物(FLP)多147Da的杂质。
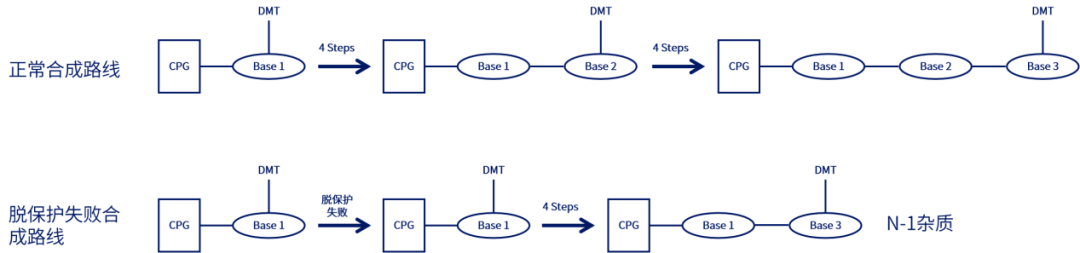
▲脱保护失败导致N-1杂质示例
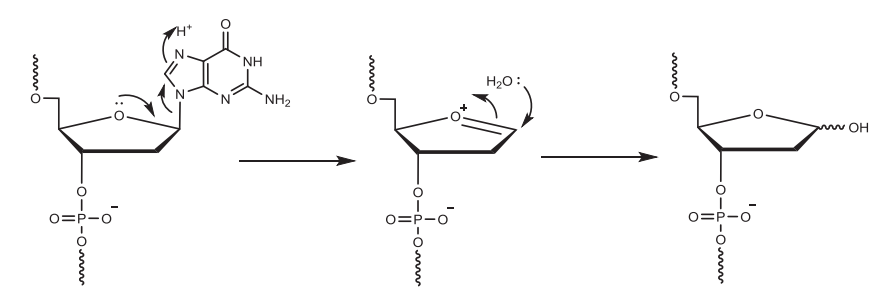
▲酸性条件下脱嘌呤的机理
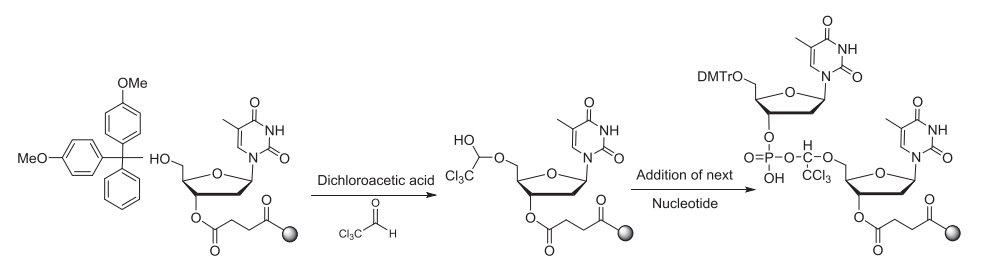
▲DCA中的三氯乙醛造成N+147杂质
02.偶联(Coupling)
下一个核酸的亚磷酰胺单体与四唑反应生成活性中间体(此步骤也被称为活化)。脱保护中形成的5'-OH亲核进攻活性中间体,形成新的磷氧键,并脱去四唑,核苷酸链得到延伸。为了保证较高的总产率,每个循环中都需要有较高的偶联效率。N-1杂质是偶联中最常见的杂质,它们是偶联效率低于100%的结果。对25-mer寡核苷酸的N-1杂质用dA延长尾巴,与有dT尾的质粒退火克隆后随机挑选70个克隆进行测序,结果显示,其中包含了所有可能的N-1序列,但3'-端截短的核苷酸的频率(序列克隆数百分比)高于5′-端截短的核苷酸。推测这个现象是空间位阻使四步反应在3′-端的效率较低导致,3'-端短的延伸链彼此靠近,DMTr也阻碍了反应;此外,空间位阻也降低去除DMTr基团的效率,使受保护的链无法进行偶联。与FLP相比,更高分子量的杂质(例如N+1)也存在于偶联步骤中。长序列杂质的形成归因于活化剂四唑的弱酸性能移除一部分亚磷酰胺溶液中的DMT基团。有两种机制导致这种杂质的形成,一种是新加入的亚磷酰胺单体自身形成二聚体,随后与脱保护的5'-OH反应;一种是新加入的亚磷酰胺单体被偶联两次。
03.氧化(Oxidation)
偶联反应后新加上的核苷酸通过亚磷酯键(三价磷)与CPG上的寡核苷酸链相连。亚磷酯键不稳定,易被酸、碱水解,在下一个循环的脱保护酸性环境中不稳定,因此需要被氧化成稳定的五价的磷。磷酸二酯键中的2-氰乙基保护基团可以使其在后续合成中更稳定。常用的氧化剂为碘的四氢呋喃//吡啶/水溶液。此外通过将一个硫原子转移到P(三价)上也可以将其转化为P (五价),从而形成硫代磷酸酯键。在合成同时含有磷酸二酯(PO)键和硫代磷酸酯(PS)键的寡核苷酸时,此步中使用的氧化试剂可以将已形成的PS键氧化为PO键,得到比FLP少16 Da的杂质。不完全的氧化/硫化中的亚磷酸酯键与后面脱保护形成的DMT阳离子反应产生两种对酸稳定的杂质。一种杂质比N-1多366 Da,另外一种杂质比FLP多286Da。
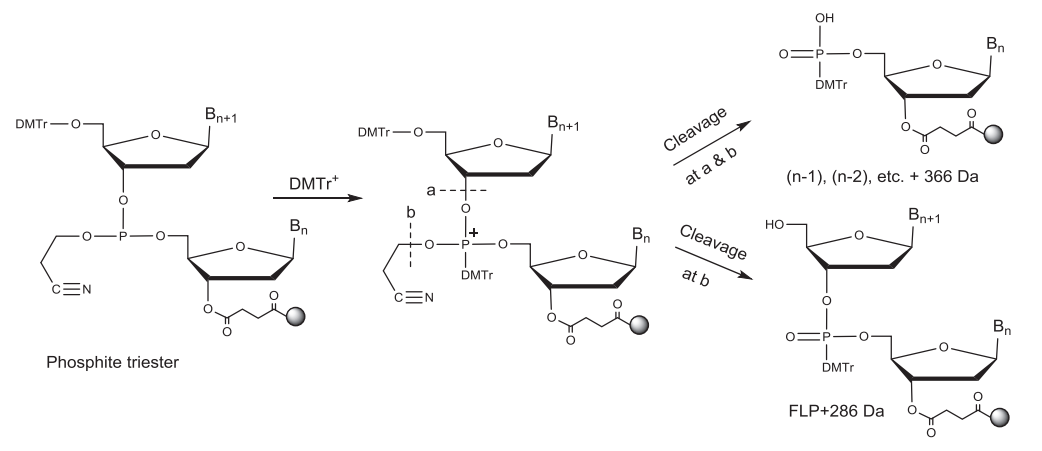
▲不完全氧化/硫化导致杂质的形成
04.加帽 (Capping)
由于不可能达到100%的偶联效率,仍存在脱保护后没有反应的5'-OH活性基团,如果不加处理,那这些基团在下一个循环中仍能发生偶联,产生N-1杂质。通常使用两种试剂(加帽试剂A,通常是醋酸酐;加帽试剂B,通常是N-甲基咪唑,酰化反应的催化剂)来酰化5'-OH。加帽步骤中混合分开存放的试剂A和B,并将其递送至系统中。乙酸酐通过将受保护的鸟嘌呤转化为乙酰化二氨基嘌呤产生比预期多41 Da的杂质。
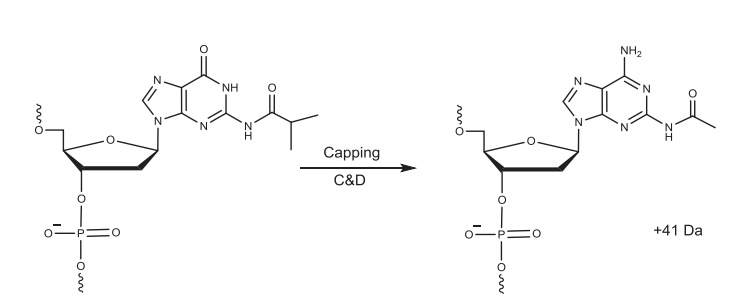
▲加帽步骤中杂质的形成
05.裂解和脱保护(Cleavage and Deprotection,C&D)
重复循环直至合成所需长度的寡核苷酸。合成结束后,可以采用氨水(或氨水/甲胺的混合物,AMA)处理将寡核苷酸从CPG上断裂并将碱基脱保护(反应速度较慢)。断裂下来的寡核苷酸末端带有游离的3'-OH。使用通用的UnyLinker固定载体时,不完全的断裂将会导致比FLP多261或275 Da的杂质的产生。在较高温度下孵育更长的时间可以将杂质转化为最终的产物。
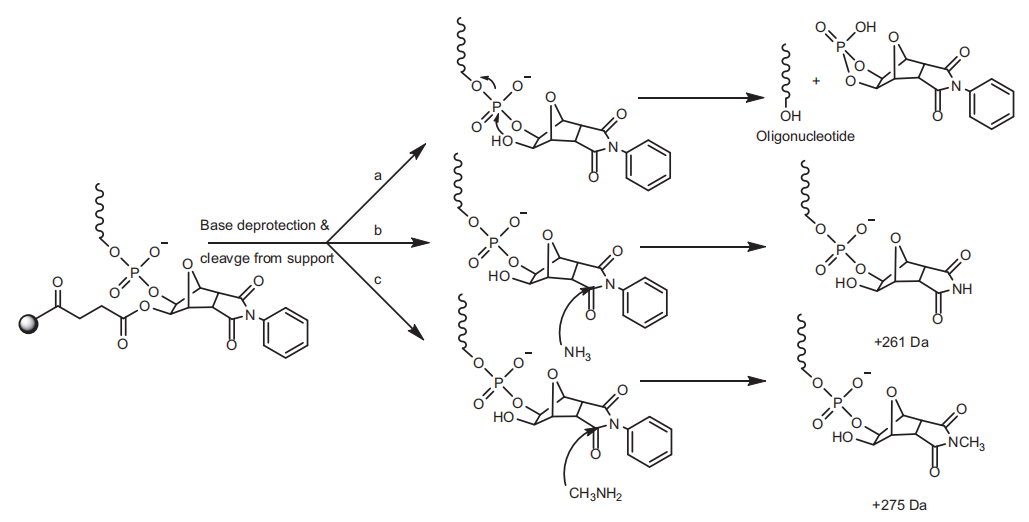
▲通用型UnyLinker固定载体上寡核苷酸与载体的不完全断裂导致杂质的产生
当寡核苷酸(RNA)中2′-OH脱去TBDMS基团保护时,游离2′-OH与载体上的羟基之间存在竞争以攻击磷,产生带3′端磷酸盐的寡核苷酸。一旦这种杂质形成,很难将它去除。采用氨水处理给碱基脱保护时,鸟苷中的异丁基保护基是最难去除的基团之一,需要在55°C孵育数小时,鸟苷富集序列则需要更长时间。鸟苷的不完全脱保护会产生比FLP高70 Da的杂质。AMA的脱保护减少了反应时间,但增加了产生比FLP高14 Da的杂质的可能。此外,甲胺可以与胞苷中的C-4反应生成N-4-甲基胞苷,在胞嘧啶上使用乙酰保护基可以减少这种杂质。鸟苷去保护所需的更严格的条件也会导致PS转化为PO,需要优化孵育时间(时间取决于序列),在鸟苷完全去保护的同时保持最小比例的PS到PO的转化。C&D的碱性条件可以发生脱氨基反应,将胞苷和腺苷的环外胺转变为碳基。非酶脱氨基由酸碱催化,并在高温下加速,脱氨前后质量差只有1 Da。
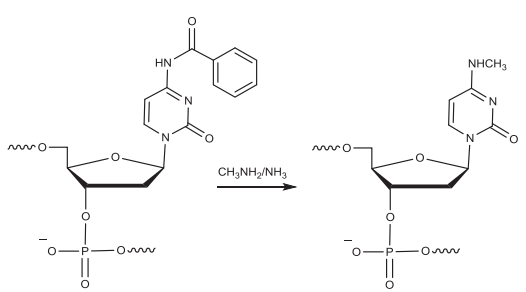
▲在断裂和脱保护过程中,甲胺与胞苷残基(被苯甲酰基保护)反应形成N-4-甲基胞苷杂质
在2′-OH采用TBDMS保护时,C&D阶段TBDMS的不完全去除产生比FLP高114 Da的杂质(RP-HPLC中比FLP晚出峰)。通常用有机盐中的氟化物离子如TEA-3HF等处理进行脱保护。TEA-3HF的酸性能中和C&D溶液的碱性,否则2′-OH暴露在碱性条件下很可能导致链断裂。这会导致环状磷酸盐的形成、5′-2′异构化(可用色谱从FLP中分离出来)和3′端截短的序列。
使用甲胺的RNA的C&D中,甲胺与尿嘧啶发生反应导致比FLP小52 Da和95 Da杂质的产生。
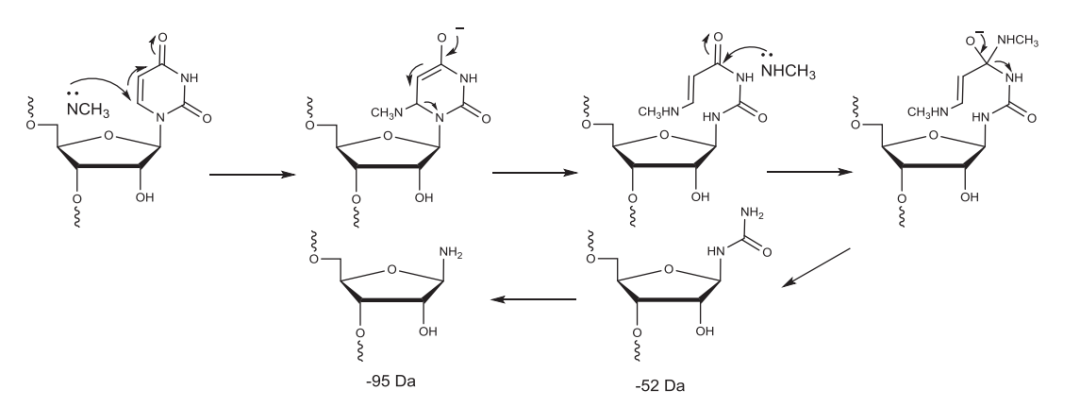
▲甲胺与尿嘧啶反应示例
2′-F修饰的寡核苷酸在经历苛刻的C&D条件时会导致HF的丢失,随后水分子的添加最终导致2′-F被羟基取代,并在2′位上发生立体化学翻转。
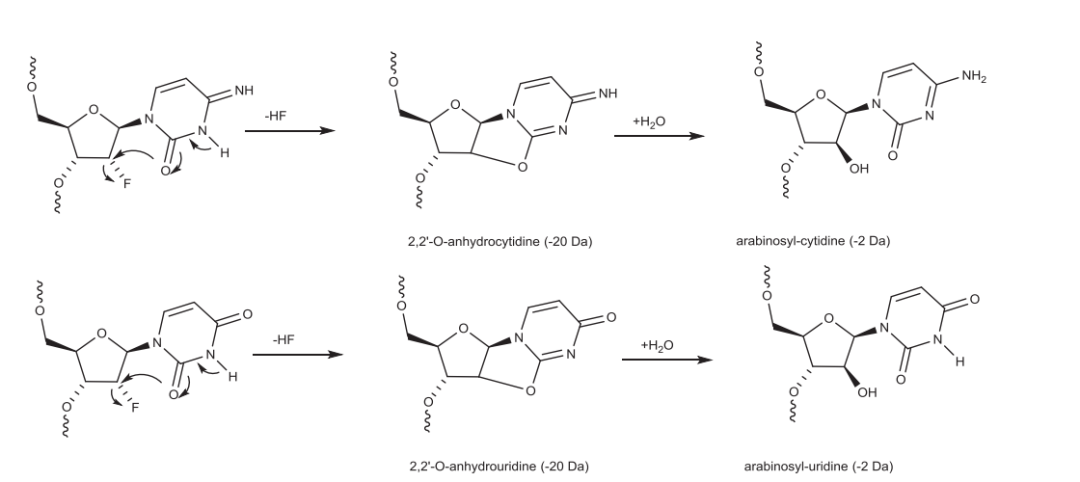
2′-F胞苷和2′-F尿苷中HF的消除
在随后的纯化阶段,多使用离子交换层析、反相层析去除产物中的杂质。经纯化后的寡核苷酸可以长期保存或直接用于后续分析。
二、寡核苷酸的修饰
未经修饰的寡核苷酸进入人体后容易被核酸酶降解,并显示出不利的细胞摄取和生物分布。寡核苷酸上通常被修饰的位置有磷酸骨架(如硫代磷酸酯键PS等)、核糖(如2′-OMe、2′-F以及核酸糖LNA等)、嘧啶碱基(如5′-甲基胞嘧啶等)以及靶向配体(如GalNAc等)的添加。研究表明,核糖的2'化学修饰(如2′-OMe、2′-F以及2′-MOE等)能降低核酸酶降解小核酸药物的能力,同时也显著降低了TLR-3/7/8识别小核酸药物为外源性核酸的能力;并大大提高了核酸的稳定性和整体半衰期。PS修饰虽然些微降低了小核酸和靶序列结合的亲和力,但它提高了寡核苷酸在血液和组织中的核酸酶抗性。由于氧原子被硫原子取代额外引入了一个手性中心,每个PS键上存在两个立体异构体(Rp结构和Sp结构)。当一种构象可被核酸酶降解时,另一种构象往往表现出较强的核酸酶抗性。同时,PS的电子云往往比PO更加分散,从而导致更多的疏水性。这一性质起到了促进蛋白质结合并延长血浆半衰期的作用,但非特异性的蛋白质结合能力往往也导致毒性,因此PS修饰的数量往往在成药性研究中被严格控制。LNA以及cEt和tc-DNA修饰涉及糖环的桥接,它们各自促进RNA样结构,显示核酸酶抗性,最重要的是,显著增加了和靶序列结合的亲和力。
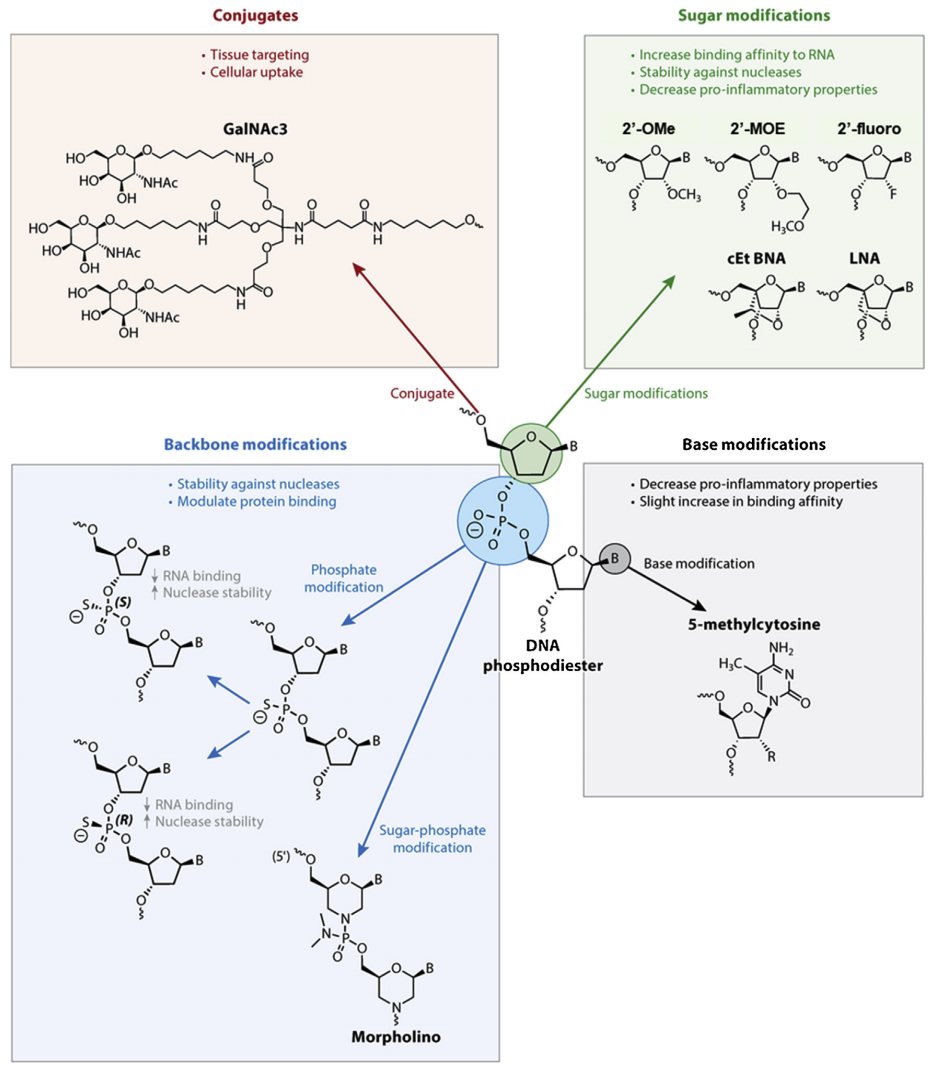
▲寡核苷酸的常见修饰
三、寡核苷酸的质量分析
目前寡核苷酸质量分析方法主要有HPLC、LC-MS、ELISA、RT-qPCR、CGE等,下图展示了文献中对单链、双链和偶联后的寡核苷酸活性药物成分(API)的检测项目和分析方法。
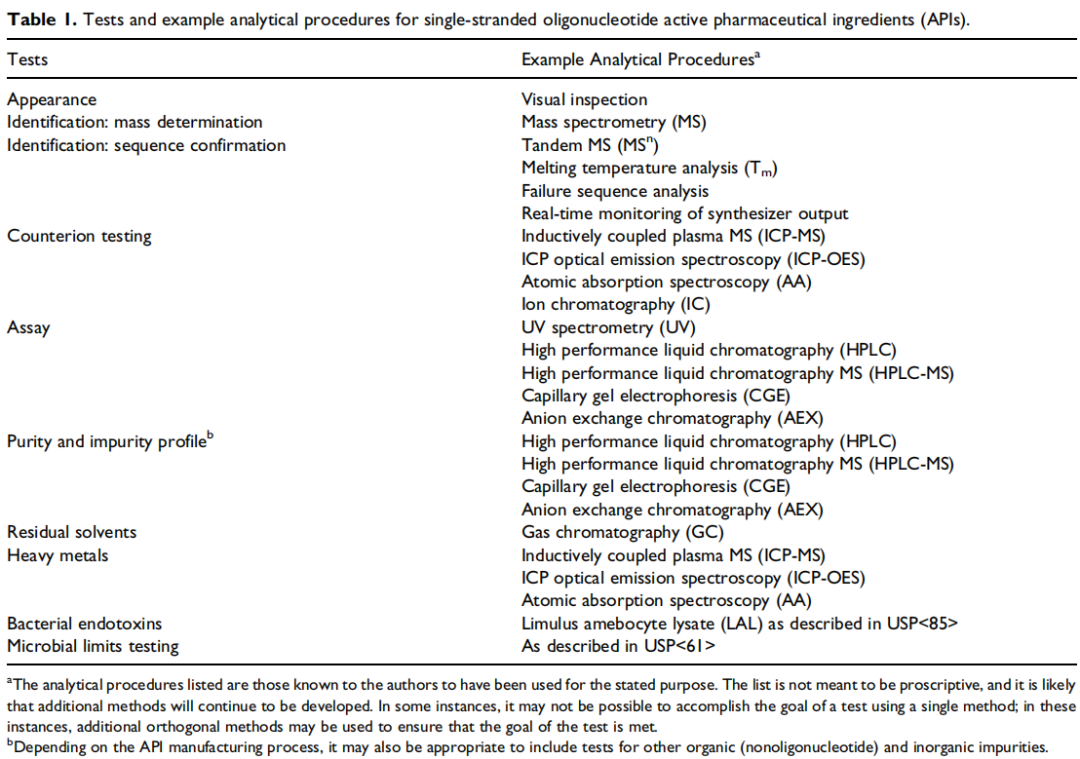
▲单链寡核苷酸活性药物成分(API)的检测项目和分析方法示例
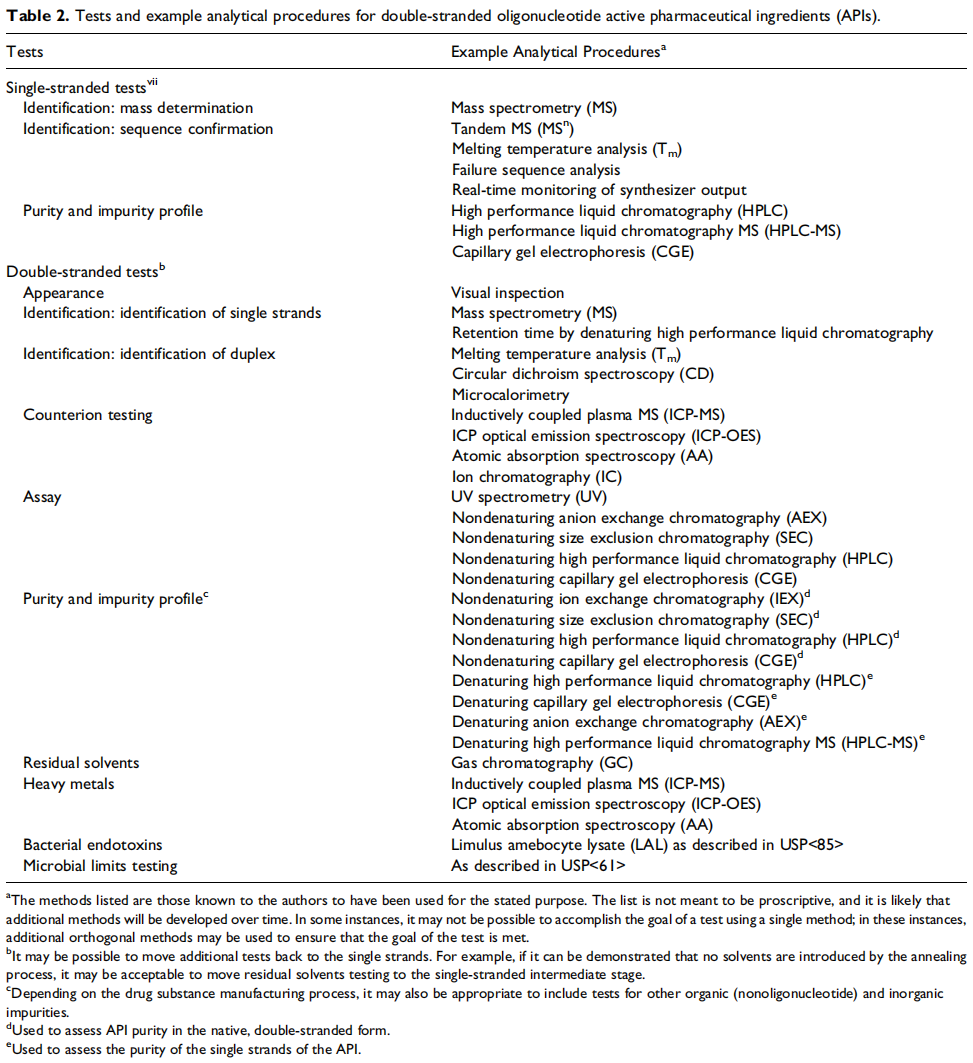
▲双链寡核苷酸活性药物成分(API)的检测项目和分析方法示例

▲偶联后的寡核苷酸活性药物成分(API)的检测项目和分析方法示例
本次小编主要针对上述亚磷酰胺固相合成产生的杂质介绍LC和LC-MS在寡核苷酸质量分析中的应用。
01.对PS键产生的立体异构体的分析
PS键在磷处引入了一个手性中心,加上D-核糖的手性中心,每个PS键产生两个非对映异构体。对于完全硫代的具有20个PS键的21-mer的寡聚核苷酸,将产生一百多万个异构体(220= 1,048,576),这对寡核苷酸的质量表征造成了非常大的挑战。通过合成包含524,288种立体异构体的反义寡核苷酸药物mipomersen,证明了Sp形态的PS键相对Rp更稳定,能够提供立体化学保护,防止药物的药理失活。采用变性IP-RP-UHPLC分离含有三个PS键的双链siRNA(正义链1个PS,反义链2个PS,双链含有8种立体异构体,变性后有6种单链立体异构体),通过优化柱温(80℃,C18色谱柱,高于siRNA退火温度的柱温有利于立体异构体的分离)、离子对试剂(0.1 M TEAA)、流动相中有机溶剂的选择(ACN,可以有效地破坏siRNA双链的自结合)、流速、洗脱梯度使6条链得到了基线分离。
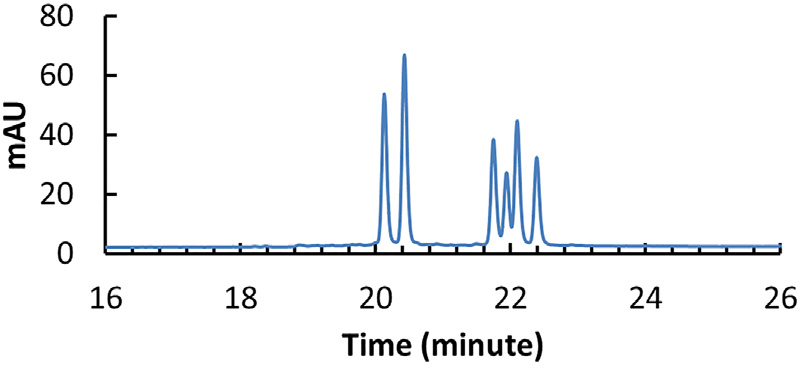
▲实验条件优化后得到基线分离的6个立体异构体
02.对PS脱硫后产生的PO杂质的分析
在含有PS修饰的寡核苷酸中,PS键可能发生氧化形成PO基团,从而影响药物的稳定性。
有报道表明,在使用ESI(-)质谱进行寡核苷酸表征时,常规离子源条件下有意外的脱硫杂质产生,此种脱硫是羟基自由基(电晕放电诱导产生)引起的S被O的取代造成,脱硫的比例取决于硫代寡核苷酸与羟基自由基的的摩尔比。此外,HPLC流速增加、硫代寡核苷酸柱上浓度的增加、电喷雾电压的降低(减少电晕放电)会降低脱硫的比例,后者在分析低水平PO杂质时会显著降低灵敏度。
03.对脱氨引起的杂质的分析
脱氨基主要发生在胞嘧啶(C)和5-甲基胞嘧啶残基上,随后得到尿嘧啶(U)和胸腺嘧啶(T),在IP-RP-HPLC分析中通常与磷代寡核苷酸的的主峰共洗脱。由于只比FLP大1 Da,如果在色谱上没有和主峰得到分离,则很难用质谱进行定量。即使在高分辨率的质谱中,杂质和主成分的同位素轮廓也会重叠,导致其定量更加困难。下图(A)所示为寡核苷酸和脱去一个氨基的降解产物在高分辨质谱中脱去四个质子时的同位素轮廓叠加图,1 Da的质量差只导致了同位素轮廓小于0.005 Da的差异。FLP中丰度最高的峰m/z为1790.0021,而脱氨基后的杂质丰度最高的峰则为1790.2481。将FLP和杂质以9:1混合后得到下图(B)所示的质谱图,其中最高信号峰(P5)之前的峰(P1-P4)相比FLP本来的强度有所降低,而P5之后的峰的强度则得到了增加,因此利用两种物质(FLP和其脱氨产物)同位素相对强度的差异可以估计脱氨基寡核苷酸的相对含量。
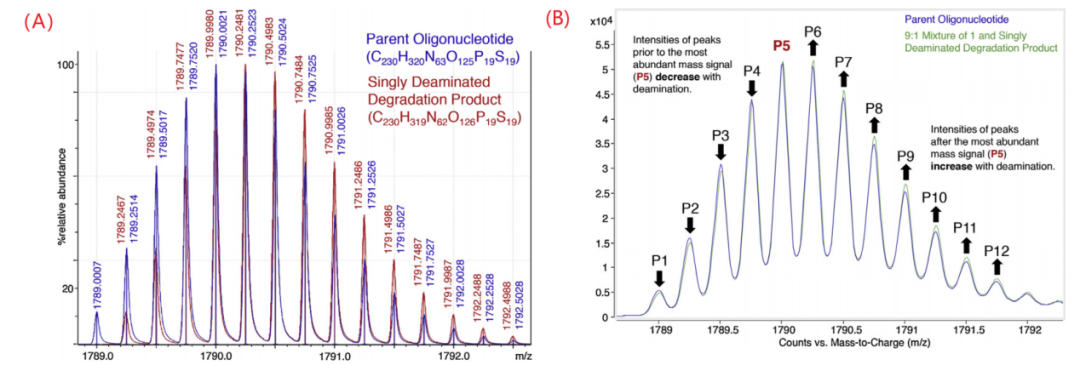
▲寡核苷酸和脱去一个氨基的降解产物在高分辨质谱中脱去四个质子时的同位素轮廓叠加图,1 Da的质量差只导致了同位素轮廓小于0.005 Da的差异
四、小 结
除了上述介绍的几种杂质,N-X、N+X型杂质的分离检测也得到了广泛的研究。2'-5'连接异构型杂质被报道可以从3'-5'中在色谱上得到分离。值得注意的是,在使用IP-RP-HPLC对寡核苷酸进行分析时,流动相中离子对试剂的选择及其浓度、不同固定相类型色谱柱的使用对主成分及杂质的分离都有着重要的影响,此外,离子交换色谱(IEX)也在寡核苷酸的杂质分析中被经常使用。在液相连接了质谱作为检测器时,离子源条件的设置对样本中杂质真实含量检测的影响也需要被注意,在确保没有源内裂解的同时最大化质谱响应信号。寡核苷酸在质谱碰撞室中解离得到充足的碎片离子可以帮助判断序列异构体的存在、修饰及其位点,这在未知杂质的判断中起着非常重要的作用。在对杂质进行充分了解和分析的同时,杂质的控制包括对起始原料的控制、序列及修饰的选择也是非常关键的一个环节。例如,在采用PS修饰的寡核苷酸中一定会产生PO杂质,此时选择少硫代或者不硫代则可以避免与硫代相关的一系列杂质的产生。此外,硫代率的检测也可以帮助产品质量控制。未来,随着检测技术的进一步发展,对于寡核苷酸药物的质量研究及产品和工艺相关杂质的分析控制也会越来越成熟,监管申报要求也会更加清晰明朗。
参考资料:
Mass Spectrom Rev, 2021. 40(2): p. 75-109.
Nucleic Acids Research, 1995. 23(14): p. 2754-2761.
Annu Rev Med, 2019. 70: p. 307–321
Nucleic Acids Research, 1995. 23(11):p. 1841-1844.
Nat Biotechnol, 2017. 35(9): p. 845-851.
Drug Information Journal, 2012. 46(5): p. 611-626.
Journal of Chromatography A, 2017. 1500: p. 84-88.
J Mass Spectrom, 2012. 47(7): p. 836-44.
J Pharm Biomed Anal, 2019. 173: p. 56-61.
Anal Biochem, 2011. 414(1): p. 47-57.
<END>




















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论