





























药物开发和生产的各个阶段的根基是分析方法。分析方法需要开发、验证和控制,就像所有其他产品和过程开发活动一样。原料药特性、影响API性质的因素和关键杂质的检测是药品研发有效性和安全性的核心。
从药品研发历史上看,含量对产品质量、放行检测和产品控制的关注严重不足。用于测定精密度的变异系数(Coefficient ofVariation, CV)计算是必要的,但不足以衡量测定方法的可靠性,并且可能因为CV与产品验收和放行检测限度没有关系而具有误导性。
分析和测定系统必须看作是一个过程。检测过程由方法、软件、试剂、分析人员、样品制备、环境条件和仪器/设备组成。质量风险管理和统计数据分析技术应用于整个实验过程,并识别可能影响精密度、准确性、线性、信噪比、检测和定量的限度和任何其参数的因素,以获得最佳的分析结果。
本文根据ICH指导原则中Q2(R1)、Q8(R2)和Q9指南讨论了一种分析方法开发和验证的系统方法。大致可以分为一下十个步骤。
1.确定分析方法的目的(表征/放行)和所有关键质量属性(Critical QualityAttributes, CQAs)
分析方法的目的有两种,一种是用于样品的放行检测一种是用于工艺过程中的控制。根据ICH Q8(R2)和样品特征确定的终产品的CQAs。对于杂质控制来讲,需要控制的杂质范围,杂质的限度,若杂质不做控制的风险评估,该方法与其他方法相关性,每个测定方法与用于评价产品的其他测定方法的正交性如何。在药物开发和生产过程中,该检测方法如何最小化或影响风险。
2.根据CQAs和开发目标选择合适的分析方法
分析方法有很多中,最需要关注的是方法需要具有较高的选择性和可靠性。
3.确定与方法相关的流程步骤
确保所有的分析方法步骤,并且详细说明了流程、材料试剂和仪器的使用。确定实验中可能产生偏差和精确度的步骤。
4.确定所有与发布测试相关的规格限制
对于那些将用于放行测试的分析方法,控制产品放行的限度是什么? 限度设定的依据可以从历史数据,行业标准,基于多批次数据统计学的偏差和平均值等确定。限度需要反映患者用药风险、CQAs保证和药品生产过程中的物料和终产品的控制。
5.对需要进行分析的地方进行风险评估
分析方法风险评估用于确定分析的步骤对精密度、准确度、线性、专属性、灵敏度(信噪比)等的影响。可以使用故障模式与理象分析(Failure Mode andEffects Analysis , FMEA)或其他风险评估方法评估。此外,我们需要在风险中添加CQAs和不确定性的影响因素进行排名。验证过程均应从这个角度出发,对具体可能影响精密度或者准确度的因素进行验证。
6.a.系统设计(合适的工艺/设备/技术)
b.参数设计(设计范围)
c.可接受范围设计(允许偏差)
在风险评估的基础上,取样量和取样方法的确定是关键的考虑因素。
分析开发可分为三个步骤:1)系统设计,2)参数设计3)可接受范围设计。
系统设计是确保我们选择合适的材料,合适的技术和设备。参数设计是通常是通过DOEs来完成的,并确保方法在最佳设计范围。可接受范围设计中的精密度和准确度是最为关键的两个指标。
7. 完成验证
图1方法验证项目汇总
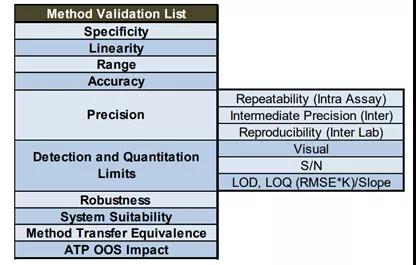
图2方法验证操作细则
 图片
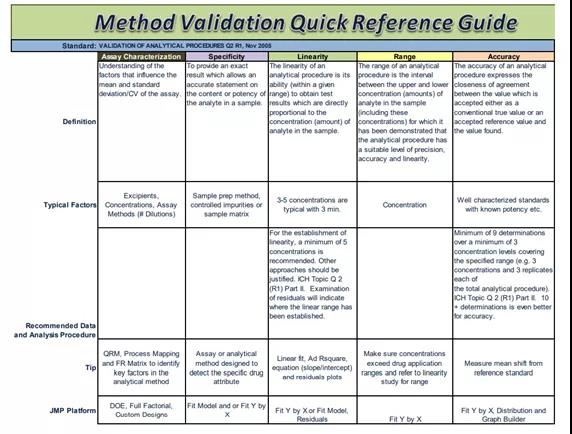
图片图3 ICH Q2(R1)中分析方法验证内容

8.定义方法的控制策略
一旦该分析方法完成开发和验证,就需要制定一个明确的控制策略(PAT)。例如:被分析物和参比分别是什么?如何确定参比的稳定性?什么将用于跟踪和趋势分析药品随时间推移发生的变化?一旦检测结果发生漂移,用什么来调整/修正?多套数据资料见如何互相参引和利用等。
9.对所有分析人员进行该方法的培训
应对所有使用分析方法的人员进行培训。分析方法中可能出错的点有:样本选择,样品制备、称量、混合、稀释、浓缩、峰位置、进样方法、时间变化等。
10.确定分析方法对工艺变更、验证和产品合格率的影响
总体标准偏差可以表示为:

随着分析方法误差的增加,总体误差也随之增加。利用准确度到精密度(Precision ToAccuracy , ATP)模型,可以可视化产品合格率的准确度和精密度之间的关系。ATP模型显示了精密度和准确度的变化如何影响产品合格率和分析误差设计空间。一般使用数字5.15表示99%的分析误差。总体测量误差%= (标准偏差测量误差*5.15)/(限度最高值-限度最低值)。
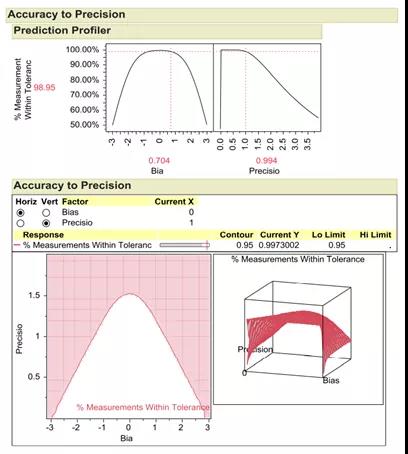
图4精密度和准确度的关系
重视方法的开发、验证和风险控制将极大地提高方法的有效性、药物研发质量、患者安全。












 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论