































1 研究背景
据药融云数据库显示,2015年1月30日,美国FDA批准新型复方降糖药物Glyxambi(恩格列净/利格列汀)的上市申请,用于2型糖尿病成年患者的辅助治疗。该药也是目前美国唯一的一款SGLT2抑制剂(恩格列净[empagliflozin],勃林格殷格翰)和DPP-4抑制剂(利格列汀[linagliptin],礼来)的复方药物。
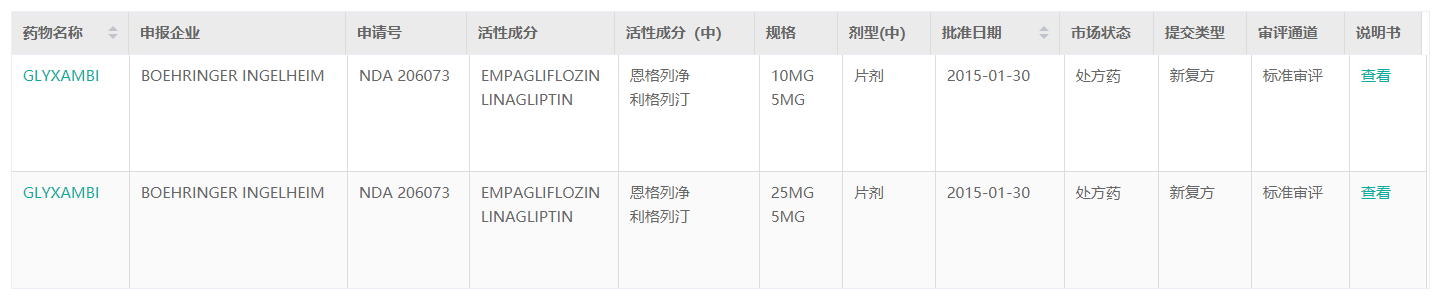
截图来源:药融云全球药物研发数据库
恩格列净利格列汀片(Ⅰ)(Ⅱ)含有两种活性成分:恩格列净和利格列汀(Empagliflozin/Linagliptin),(Ⅰ)每片含恩格列净10mg和利格列汀5mg;(Ⅱ)每片含恩格列净25mg和利格列汀5mg。
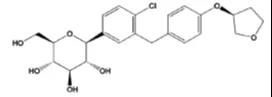
恩格列净化学结构如下:
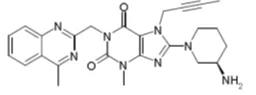
利格列汀化学结构如下:
根据2020版中国药典四部通则0941 含量均匀度检查法可知,除另有规定外,片剂、硬胶囊剂、颗粒剂或散剂等,每一个单剂标示量小于25mg或主药含量小于每一个单剂重量25%者,均应检查含量均匀度。本品为复方固体口服制剂,含恩格列净和利格列汀两种活性成分,其制剂包括10mg/5mg和25mg/5mg两种规格(其中10mg和25mg均为恩格列净规格、5mg为利格列汀规格),片重均为185mg,即两种规格制剂的两个主药含量均小于每一个单剂重量25%,应检查其含量均匀度,考察单剂量的固体制剂含量符合标示量的程度。
2 方法开发思路
本品含量均匀度检测方法参考原料药和单方制剂含量检测方法,复方有关物质检测方法以及溶出度检测方法色谱条件,初步选定苯基柱检测两种主成分含量,计算方式采用单点外标法(两个规格,即采用两个对照),其色谱条件的筛选、前处理条件以及溶液配制的确定,详见下文。
2.1 色谱条件的确定
(1)研究过程
分别称取适量恩格列净和利格列汀对照品,置于同一容量瓶中,加入适量稀释溶剂(pH2.5磷酸盐缓冲液:乙腈=70:30)使溶解,制成每1ml中约含利格列汀0.010mg和恩格列净0.050mg的混合对照品溶液。精密量取上述溶液10μl注入液相色谱仪,分别按下表色谱条件进行样品分析,记录色谱图。
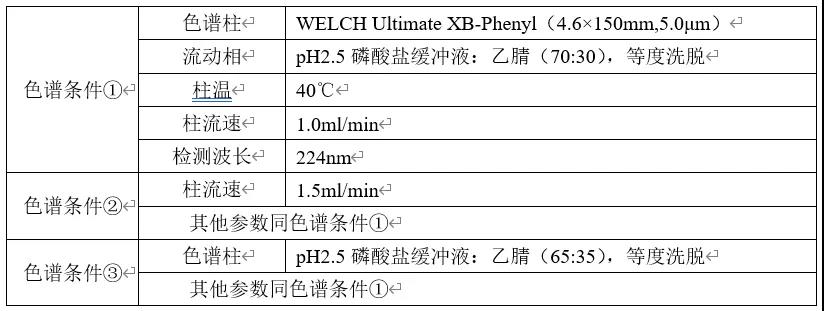
(2)结果与讨论
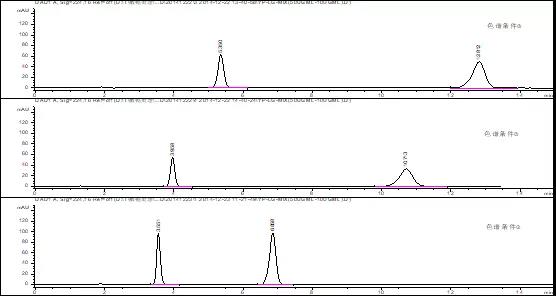
由以上色谱图可知,1)增加苯基柱可明显调整各主成分的保留时间,因①条件下两成分出峰时间较晚,故尝试通过增加柱流速或提高有机相比例的方式,缩短整体的分析时间;2)增加柱流速对于恩格列净色谱峰型和保留时间没有明显改善;3)色谱条件③下,利格列汀出峰时间为3.551min(对称因子0.83),恩格列净出峰时间调整为6.858min(对称因子1.11),出峰时间差△TR<3.5min;4)目前各成分峰高较为合适(均在100mAU,推测其恩格列净浓度为0.020mg/ml时响应峰高约为50mAU),故可暂定为恩格列净利格列汀片含量检测方法色谱条件。
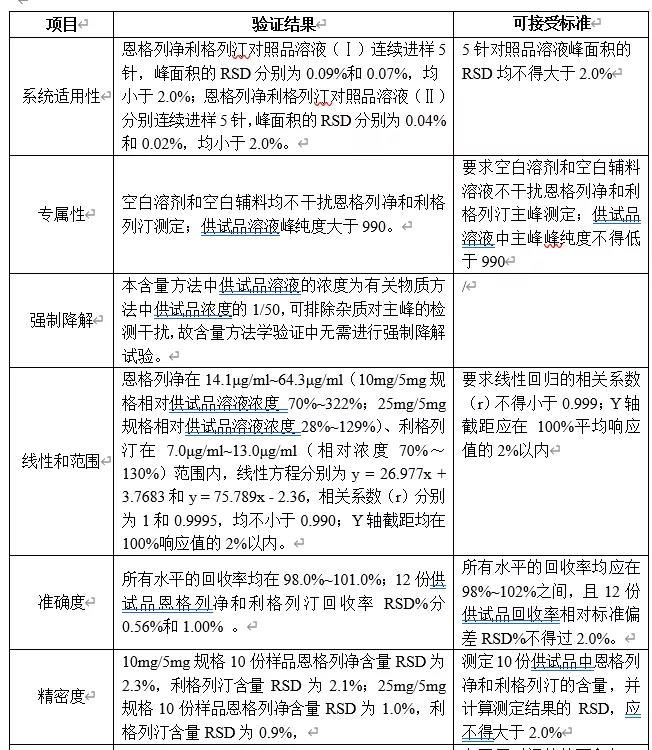
对暂定恩格列净利格列汀片含量检测方法的色谱条件进行简单验证(系统适用性、专属性和线性),结果见下表。
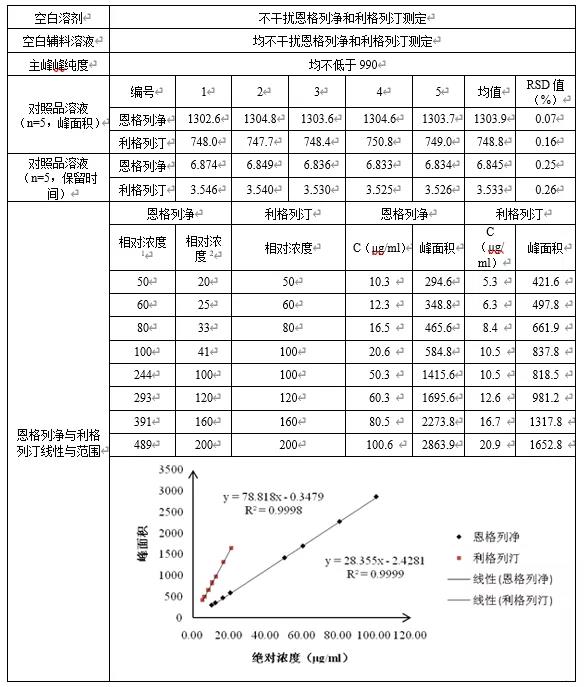
- 相对浓度1:相对10mg/5mg规格恩格列净供试品溶液(20μg/ml)的相对浓度;
- 相对浓度2:相对25mg/5mg规格恩格列净供试品溶液(50μg/ml)的相对浓度;
- 相对浓度:相对利格列汀供试品溶液(10μg/ml)的相对浓度
由上表可知,1)本法系统适用性和专属性均可满足要求;2)10mg/5mg规格制剂供试品溶液浓度定为恩格列净0.020mg/ml、利格列汀0.010mg/ml,25mg/5mg规格制剂供试品溶液浓度定为恩格列净0.050mg/ml、利格列汀0.010mg/ml,本法在恩格列净0.010mg/ml~0.1mg/ml(相对10mg/5mg规格供试品溶液浓度为50%~489%、相对25mg/5mg规格供试品溶液浓度为20%~200%),利格列汀0.005mg/ml~0.020mg/ml(相对浓度50%~200%)范围内,线性较好,故可作为恩格列净利格列汀片含量检测方法色谱条件。
2.2 前处理条件的确定
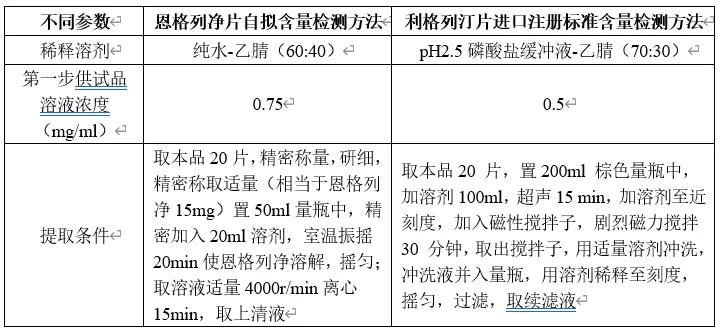
本品处方组成与单方制剂利格列汀片较为接近,推测其辅料干扰和提取难度较为接近;另对比恩格列净片自拟含量检测方法前处理条件和利格列汀片进口注册标准含量检测方法前处理参数如下。
由上表参数推测本品中利格列汀提取难度大于恩格列净,故为考察提取参数(为简化操作,将超声后磁性搅拌调整为室温下振摇)考察本品含量提取超声或振摇时间,考察结果见下表。
由上表可知,确定本品含量测定前处理条件为超声15min,再室温振摇30min。由上表参数推测本品中利格列汀提取难度大于恩格列净,故为考察提取参数(为简化操作,将超声后磁性搅拌调整为室温下振摇)考察本品含量提取超声或振摇时间,考察结果见下表。另对其滤过验证试验进行考察,结果如下。
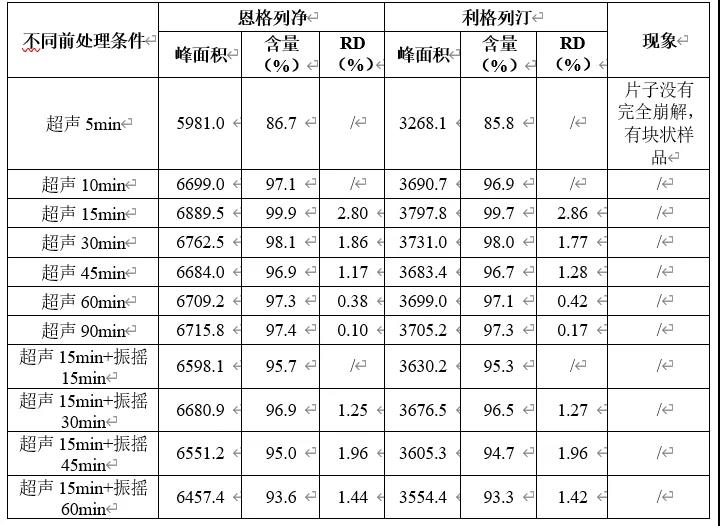
由上表可知,过滤不同体积制备所得供试品溶液与离心制备所得供试品溶液测得各成分峰面积无明显变化,即无明显滤膜吸附。
基于以上,确定前处理条件为取本品1片,置于20ml量瓶中,加溶剂(磷酸盐溶液[取无水磷酸二氢钾2.0g,加水1000ml使溶解,并用磷酸调节PH值至2.5±0.1]-乙腈(70:30))适量,超声15min,再室温振摇30min,加溶剂稀释至刻度,摇匀,用0.45μm尼龙滤膜滤过,弃去初滤液,取续滤液1.0ml置于25ml量瓶中,用溶剂稀释至刻度,摇匀,即得供试品溶液。
3 检测方法
照高效液相色谱法(ChP 2020版四部通则0512)测定。
供试品溶液 取本品1片,置于20ml量瓶中,加溶剂(磷酸盐溶液[取无水磷酸二氢钾2.0g,加水1000ml使溶解,并用磷酸调节PH值至2.5±0.1]-乙腈(70:30))适量,超声15分钟,再室温振摇30分钟,加溶剂稀释至刻度,摇匀,用0.45µm尼龙滤膜滤过,弃去初滤液,取续滤液1.0ml,置于25ml量瓶中,用溶剂稀释至刻度,摇匀。
恩格列净对照品贮备液 称取恩格列净对照品约10mg,精密称定,置于20ml棕色量瓶中,用溶剂溶解并稀释至刻度,混匀
利格列汀对照品贮备液 称取利格列汀对照品约10mg,精密称定,置于100ml棕色量瓶中,用溶剂溶解并稀释至刻度,混匀。
对照品溶液(Ⅰ) 分别精密移取恩格列净和利格列汀对照品贮备液2.0ml和5.0ml置于同一50ml棕色量瓶中,加溶剂稀释制成每1ml中约含20μg恩格列净和10μg利格列汀的溶液;
对照品溶液(Ⅱ) 分别精密移取恩格列净和利格列汀对照品贮备液2.0ml置于同一20ml棕色量瓶中,加溶剂稀释制成每1ml中约含50μg恩格列净和10μg利格列汀的溶液。
色谱条件 用苯基键合硅胶为填充剂[如WelchUltimate XB Phenyl柱(4.6×150mm,5.0μm)];以磷酸盐溶液[取无水磷酸二氢钾2.0g,加水1000ml使溶解,并用磷酸调节PH值至2.5±0.1]-乙腈(65:35)为流动相;流速为1.0ml/min,柱温为40℃,检测波长为224nm,进样体积10μl。
系统适用性要求 对照品溶液(Ⅰ)(Ⅱ)重复进样5次所得依帕列净和利格列汀峰面积的相对标准偏差均应不大于2.0%
测定法 精密量取对照品溶液(Ⅰ)(Ⅱ)和供试品溶液,分别注入液相色谱仪,记录色谱图。按外标法以峰面积分别计算恩格列净列净和利格列汀的含量,应符合规定(ChP 2020版四部通则0941)。
4 方法验证
为保证恩格列净利格列汀片含量均匀度检测方法的准确性和可靠性,根据ChP2020版四部通则9101《分析方法验证指导原则》和ICH Q2(R1)的指导原则,制定方法学验证方案,对恩格列净利格列汀片含量检测方法进行方法学验证,验证结果如下表所示。

5 多批次样品检测结果
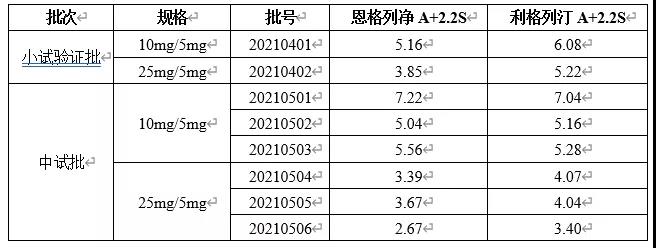
根据药典要求,取各批供试品10个,照上述含量测定法,分别测定每一个单剂以标示量为100的相对含量,求其均值和标准差S以及标示量与均值之差的绝对值A。若A+2.2S≤15.0,则供试品的含量均匀度符合规定。检测结果见下表。
6 结论
基于以上研究可知,该方法通过专属性、线性、回收率、方法重现性及溶液稳定性等方法学验证试验,显示方法可行,多批次样品检测结果均符合药典标准。
<END>

















 川公网安备51019002008863号
川公网安备51019002008863号 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论