






























一、方法验证的趋势
WHO、FDA、USP 和ISO 17025 对于分析方法验证的定义和解释基本一致,其核心是实验室通过试验设计和测试,证明被验证的方法适用于该方法拟定的检测用途。分析方法要求具有专属、准确、耐用、重现性好的特点,不同的实验室对方法验证的具体做法和可接受标准往往是不同的,但是,基本都会根据官方指导原则或药典通则规定进行方法学验证,并符合GMP要求。
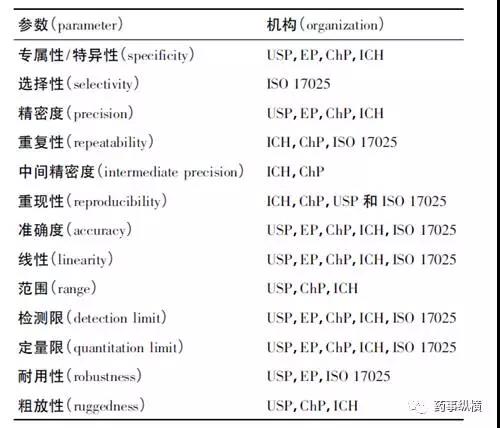
不同国际组织和药典要求的方法学验证参数
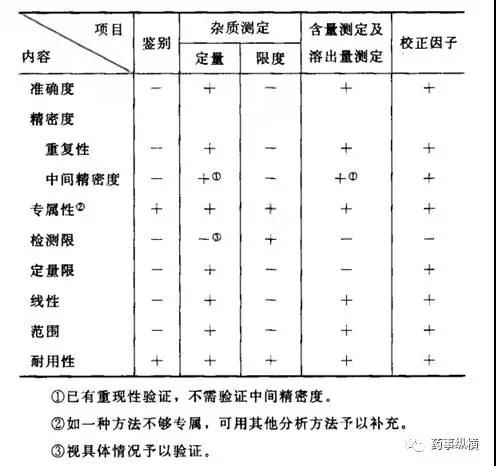
需要注意的是,2015版中国药典通则9101《药品质量标准分析方法验证指导原则》中增加了“校正因子”的验证,详见下表。
ChP2015检验项目和验证指标
CDE电子刊物《HPLC法校正因子研究中的几个问题》一文记载,在一般情况下,校正因子可视具体情况通过如下几种方法测定:
(1)单浓度点测定
(2)多浓度点测定
(3)标准曲线法测定
(4)吸收系数比值法
其中,(1)和(2)法较为简捷,可以快捷地量化特定杂质与主成分紫外吸收特征的差异,多用于评估采用主成分自身对照法定量杂质时是否需要校正。但如采用加校正因子的主成分自身对照法定量杂质,需将标准物质赋值信息转化为校正因子固化在质量标准中,那么校正因子的准确性非常关键,校正因子的准确计算应符合更为严格的要求,需要考虑并控制求算校正因子过程中的各种误差因素,以及仪器通用性和色谱系统的耐用性等因素,以便使求得的常数更为准确并具代表性,此时采用(3)、(4)法更为适宜,如能考虑到测定人员、不同试验室因素的影响,会更加符合常数求算的基本要求。
通过上述介绍不难发现,方法学验证不仅仅是完成某个指标(如回收率、重复性)的验证。指导原则或药典没有规定每个具体验证指标的试验方法,原料药、制剂、剂型、正相色谱、反相色谱、甚至不同公司SOP规定的不同都会引起结果的差异和准确性。例如回收率,有时会出现由于实验设计方法不同、对指导原则或药典理解的不同而产生不同的试验结果,或符合要求,或不符合要求,而究竟哪个是“科学合理”的,仁者见仁智者见智。
实验设计(方案)的多样性、对样品从取样到检测完毕这个过程的研究深度、是否对检测过程中的“异常”问题进行深入分析等,往往会对最终测定结果的准确性产生影响甚至会致使“超标”现象。例如:文章《辛伐他汀片的含量测定中研磨对含量结果的影响》报道,日常检验中发现辛伐他汀片同一批成品的含量与同批颗粒的含量偏低4%左右。从颗粒到压片的过程,从理论上讲不应该出现主药含量下降的情况,并且检验过程无误。由于在检验过程中颗粒和成品的区别只有研磨时间和难易程度不同,所以从研磨方面查找原因。结果表明,辛伐他汀片在含量测定过程中,取20片研磨称取细粉测定含量的结果与不研磨而是直接称取整片测定含量的结果比较相差较大。研究发现研磨程度和研钵大小都会有一定影响,所以应取用相同材质和同样大小的研钵,并且用力均一。研磨样品应都取20 片,防止样品的研磨量不同引起误差。5mg规格辛伐他汀片应直接称取4 片进行测定。部分代表性数据如下:
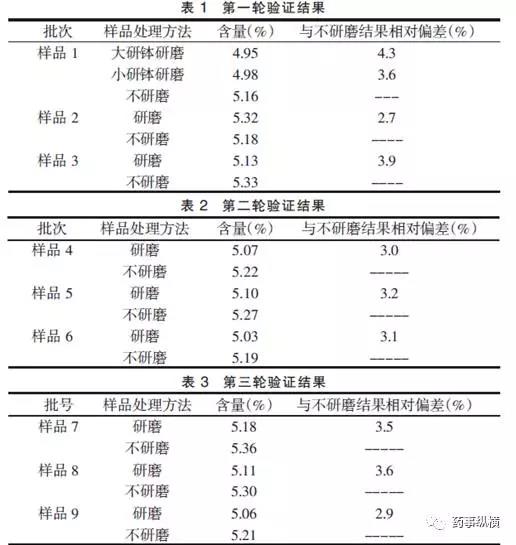
所以,在方法建立和方法验证阶段,采用简单、传统的研究思路和实验设计可能不能保证日后的测定结果的准确性和使用目的,我们需要在做验证时考虑更加细致,通过大量的比较研究、科学地去设计方案、模拟实际情况,开展方法验证。
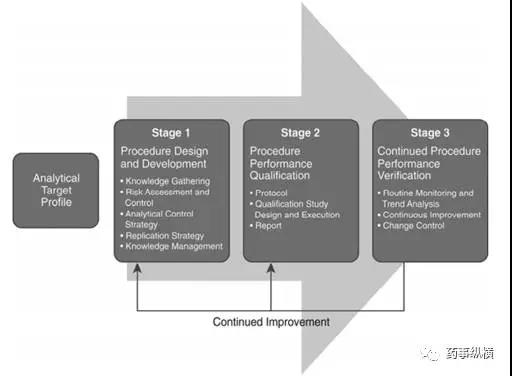
2016年10月14日,USP发布了一个新的通则“分析方法生命周期” The Analytical Procedure Lifecycle <1220>征求意见稿,并发表在美国药典论坛43(1)【2017年1-2月】上。其中的理念与ICH Q8、Q9、Q10和Q11所述的质量源于设计的理念一致。这是一个新的方法验证思路,值得我们学习和借鉴。
例如在USP <1220>中规定,将分析方法的整个生命周期划分为三个阶段。
Stage1: Procedure Design and Development (Knowledge Gathering, Risk Assessment, Analytical Control Strategy, Knowledge Management, Preparing for Qualification)
第1段:方法设计和开发(知识收集、风险评估、分析控制策略、知识管理、确认准备)
Stage2: Procedure Performance Qualification
第2段:分析方法性能确认
Stage3: Continued Procedure Performance Verification (Routine Monitoring, Changes to an Analytical Procedure)
第3段:持续方法性能确认(日常监测,分析方法变更)
分析方法生命周期中的一个基本要素是制订一个目标说明对分析方法的性能要求。这些在分析目标概况(ATP)中描述的要求可以看作是类似于产品质量目标要求(QTPP)。在ATP中,我们需要研究和控制各种不确定度,研究可能产生偏差(如专属性、线性、提取回收率、滤膜验证、检测波长、溶液稳定性、分析物的溶解度)和提高方法精密度的因素(如样品制备过程、称量、仪器、积分、背景噪音、重复次数、操作人员)。

如果采用新的研究思路,会要求应用新的实验设计。如文献《基于Q2(R1)理念的药品质量分析方法验证新概念》报道,在进行线性试验时,可根据ICH要求, 配制5种浓度水平来评估,根据eNvoal的要求, 至少需要独立的3个序列, 且每个序列至少重复3次,实验设计如下表所示。
实验设计要求目标
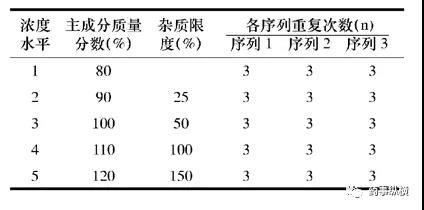
在整个实验过程中, 范围、准确度、精密度、线性和定量限被涵盖, 其中准确度和精密度能够通过eNvoal软件一次性评估。为了得到在一定置信区间内各个浓度水平某个单个测试结果的预测区间,需要通过专业的统计软件来完成。
通过eNvoal引入了准确性特征值的概念,是整个浓度范围的预测模式, 涵盖了准确性、准确性、精密度和定量限。推断预测区间和风险评估, 也可提供一般统计数据(平均值、RSD等)。浓度范围的预测的准确性和特征值,见下图。
浓度范围预测的准确性和特征值曲线图

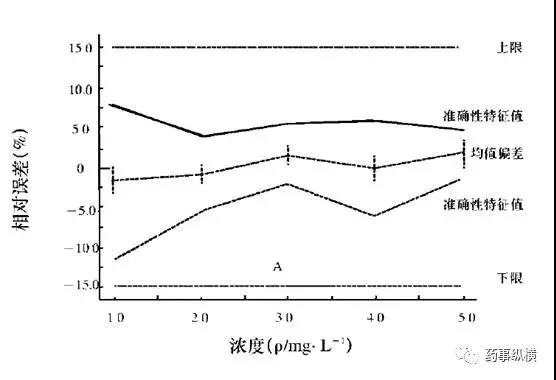
通过eNvoal能够非常容易判断出来该分析方法是否适用于检测的要求。eNvoal结果判断见下图。
eNvoal结果判断方法的有效性示意图
eNvoal结果判断方法的无效性示意图
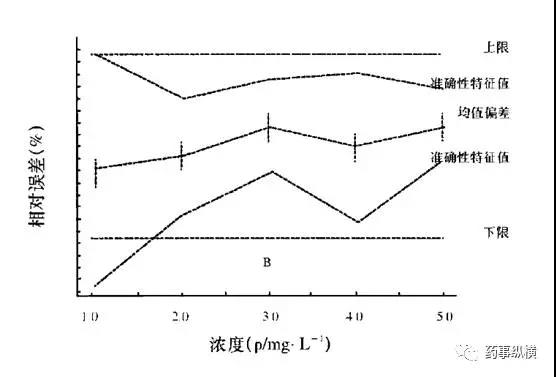
以上例子说明,与传统的验证方法相比,新的验证方法实验设计更加统一,验证后的方法更加可信,是方法验证的新趋势。
二、方法验证时需要注意的因素
对于一个分析方法需要考虑三方面因素,Sample Preparation、Measurement、Replicate Strategy。
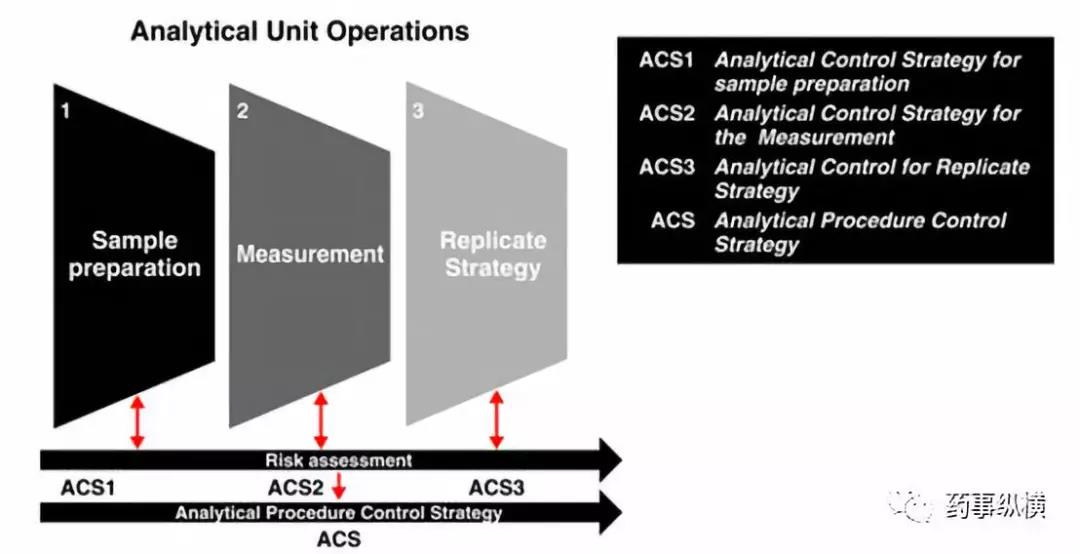
在样品的制备环节,一般包括称量、提取、稀释等环节,但是取样的代表性有时也影响结果的准确性,这种影响是不能忽视的,例如出现制剂成品含量合格,中间产品含量不合格的情况往往是由于取样过程中的问题引起的,在研究时应将这个环节进行详细分解研究。在溶液制备的过程中有两个因素很关键,一个是样品溶解/提取/溶出的是否完全,另一个是溶液的稳定性。为了保证待测成分提取完全/不被破坏,就要考虑加入的药物的量、是否存在静电吸附、溶剂体积、提取方式,提取时间、提取温度、是否避光操作等因素。
在测量这个环节,以HPLC为例,受到样品溶液、对照品溶液、仪器性能、检测器类型或能量、检测波长、检测器的参数设置、计算公式、线性范围、溶剂峰(或辅料峰)、试剂纯度、色谱柱、甚至样品小瓶等的影响,总之变量因素非常多,遇到异常问题时可以采用鱼骨图等方式进行分解研究,如下图。
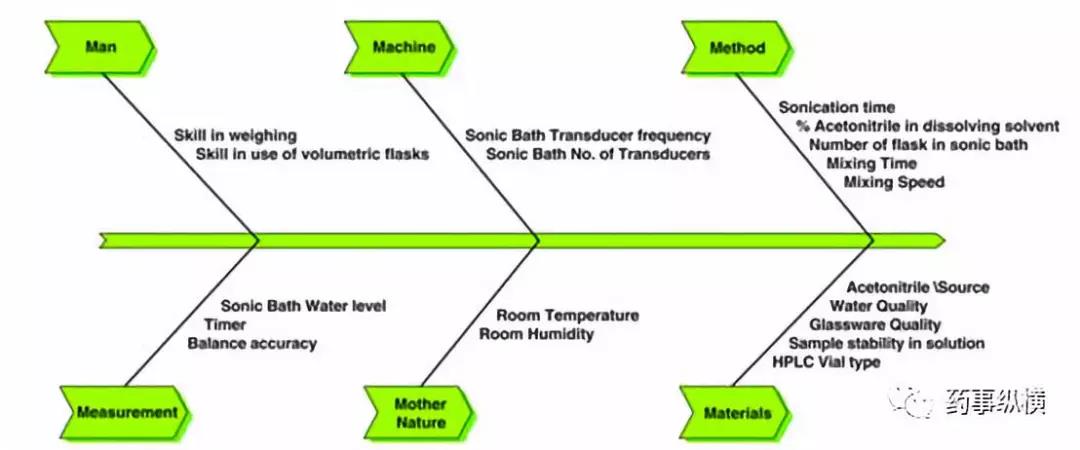
方法的重现性控制策略在方法的建立、优化、验证时就应该重要考虑,比如供试品溶液的制备方法的重现性、杂质峰相对保留时间的重现性、杂质校正因子的重现性、主峰保留时间的重现性、杂质间分离度的重现性,这些项目中的影响因素需要考虑。例如样品溶液是否受量瓶体积的影响、加入溶剂量的影响、取样量的影响;保留时间是否受到不同色谱柱的影响、相同色谱柱不同粒径或不同品牌的的影响等;分离度是否受到流速、柱温、仪器死体积的影响等。
围绕上述三个环节,列举几个方法验证中有时需要注意的因素:
1、是否有一个沟通良好的项目讨论小组。例如小组成员可包括合成、分析、制剂等。合成人员负责杂质谱的分析、杂质制备、结构确证、谱图解析、包装等;分析人员负责方法摸索、验证、杂质标定、杂质质量标准建立等;制剂人员负责提供空白辅料、相容性信息、制剂样品。大家的目的是相同的,即保证方法验证及其相关工作顺利进行,对于杂质谱分析和杂志控制策略往往需要大家一起讨论制定。
2、明确分析方法的使用目的和应用范围。方法是用于测定什么的?是测定含量、杂质、溶出度的还是做鉴别的,也就是说定量和定性的,目的不同,验证的内容也就不同。
3、明确样品类型、方法类型。样品是起始物料、中间体、反应液、原料药、还是制剂?例如对于原料和制剂来讲,二者对方法的影响是不一样的,制剂中的辅料常常会对方法造成干扰,如出现辅料峰、辅料引入的杂质峰,还要考虑辅料对主药的影响,以及溶剂提取、供试品溶液的制备方法是否合适。对于中间体、反应液等有时可能不需要准确定量、精密称取。预计使用的杂质计算方法是外标法、自身对照法、还是归一化法?
4、开展预验证试验和研究。在研究开始时分析仪器应经过DQ、IQ、OQ、PQ的确认,如对于HPLC,UV、溶出度仪进行校验,这是方法学验证的基础(如下图所示)。每次分析前进行系统适用性试验,保证系统性能符合要求。

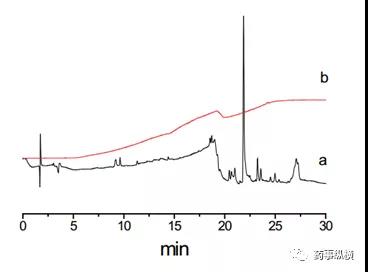
要考察对照品溶液、供试品溶液、溶剂、流动相等的稳定性情况,一般要求至少24h稳定,找到敏感因素,并控制这些因素,如温度、光等。确定系统适用性的评价方式和指标,如采用峰鉴别对照品溶液、破坏溶液、还是对照品混合溶液?采用分离度、相对保留时间、主峰保留时间、理论板数、拖尾因子,还是RSD进行评价?要通过专属性、降解试验(或混合降解溶液)等评价色谱柱,确定厂家、品牌、固定相种类等。基线波动、漂移、出现鬼峰等是梯度洗脱中常见的问题,导致在扣除溶剂峰时遇到问题。这时可以从试剂纯度、流动相使用时间、平衡时间、杂质捕集柱等方面考虑。如使用高纯度的试剂、水,带有缓冲盐的流动相现配现用,不超过48h内使用;梯度洗脱后要摸索平衡时间,选择一个合适的时间如5、10min等。当遇到鬼峰很多的情况,可以使用流动相杂质捕集小柱,如月旭科技Ghost-Buster Column流动相杂质捕集小柱安装在梯度混合器和进样器之间,不仅能够去除流动相中的杂质,还可以有效去除管路和混合器中的杂质。下图为不加 Ghost-Buster流动相杂质捕集柱(a)和加Ghost-Buster流动相杂质捕集柱(b)对比色谱图。
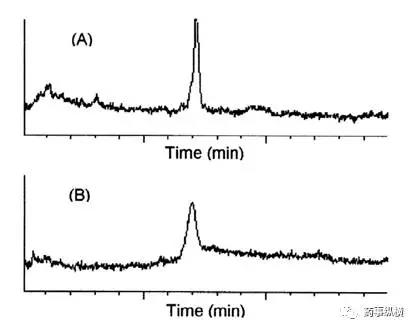
对于HPLC法的LOD和LOQ,有时受色谱图的形状影响很大,需要注意色谱柱的品牌和柱效,如下图所示。有些方法参数的研究可以尝试使用实验设计(DOE)进行研究。
5、预验证结束后可以起草一个方法的SOP和药典格式的质量标准草案,并起草方法学验证方案,尽可能详细的列出操作步骤,便于操作和重复,列出各项验证指标可接受的范围。例如列出色谱柱的详细信息、样品信息、对照品信息、仪器信息、溶剂配制详细步骤和过程、所用试剂的级别和厂家、过滤器的类型和大小等、进样顺序表等,试验中应严格遵守SOP规定。
6、方法验证结束后要及时汇总结果,整理验证报告,并经过复核和审核,将验证报告和记录等存档。但是,验证通过往往不代表后期检验样品时没有问题,仍需要对方法进行持续跟踪、完善,关注样品检测时遇到的新的问题,有时可能需要对方法进行补充研究,进行追加验证或再验证。
综上所述,药品检验的分析方法来源于合理的设计,要保证操作过程严谨科学。USP<1220>、2015年版的《FDA分析方法验证指南》、美国药典论坛中的文章如Analytical Target Profile: Structure and Application throughout the Analytical Lifecycle、Analytical Control Strategy、Lifecycle Management of Analytical Procedures: Method Development, Procedure Performance Qualification, and Procedure Performance Verification、Fitness for Use: Decision Rules and Target Measurement Uncertainty等文章对于方法建立、验证、管理具有一定的指导意义。质量研究人员可借鉴其中的思想,领会新的验证思路,同时在观察到异常现象后,主动查找原因,找到问题的解决办法,可对方法的控制策略有深刻的理解。
三、参考文献
1、分析方法验证、转移和确认概念分析
2、CDE电子刊物《HPLC法校正因子研究中的几个问题》
3、辛伐他汀片的含量测定汇总研磨对含量结果的影响
4、基于Q2(R1)理念的药品质量分析方法验证新概念
5、USP<1220> : The Analytical Procedure Lifecycle
6、USPPF: Analytical Control Strategy
<END>
















 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论